Болезнь де Тони-Дебре-Фанкони: причины, симптомы, принципы питания и лечения у детей. Синдром Фанкони: симптомы, лечение Симптомы синдрома Фанкони
Синдром Фанкони, он же болезнь Тони-Дебре-Фанкони, это состояние почек, при котором нарушается обратное всасывание подавляющего большинства веществ, что сопровождается глюкозурией (сахар в моче), аминоацидурией (белок), гиперфосфатурией (фосфаты) и наблюдаются выраженные метаболические нарушения в организме. Данное состояние у детей приводит к задержке развития, рахиту и мышечной слабости.

При синдроме Фанкони нарушается обратное всасывание веществ в почках
Причины этого состояния многообразны и до сих пор не конкретизированы. Как правило, синдром Фанкони связывают с непереносимостью , недостаточностью ряда клеточных ферментных систем (фосфоенолпируваткарбоксикиназы, например), хроническим отравлением организма токсинами, авитаминозом витамина Д.
Из-за разности в представлениях, одно и то же состояние может называться ещё и «идиопатическим ренальным синдромом Фанкони», и «Д-резистентным рахитом». Иногда встречается термин «глюкофосфаминный диабет» или «почечный нанизм с Д-резистентным рахитом».
Встречается относительно редко, что, частично, и объясняет недостаточность информации об этом синдроме. В среднем, синдром встречается у одного из 350 000 младенцев.
Разновидности синдрома Фанкони

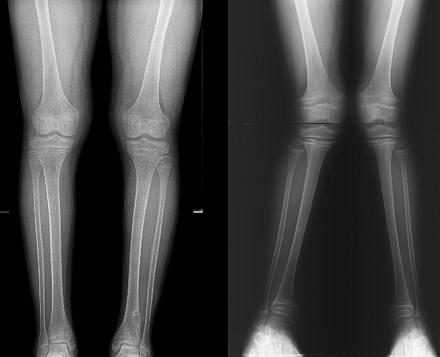
На фоне дефицита фосфатов, голени приобретают О-образную форму
Выделяют врождённый (первичный, идиопатический) синдром, и приобретённый.
Первичный синдром Фанкони считается сцепленным с Х-хромосомой.
Может быть доминантным или рецессивным, т.е. наследуется самым разнообразным путём, прогнозировать наличие у потомства достаточно непросто.
Генетически обусловленный синдром Фанкони, может быть полным и неполным, т.е. иногда проявляются лишь 2 из 3х классических симптомов (глюкозурия, аминоацидурия, гиперфосфатурия).

Первичный синдром Фанкони является генетическим заболеванием
Вторичный, как правило, являедся следствием , тирозинемии первого типа. Способствуют возникновению синдрома наследственные патологии почек. Синдром Фанкони достаточно часто развивается после органа (при низкой гистосовместимости).
Болезнь Тони-Дебре-Фанкони может быть следствием отравления ртутью, ураном, свинцом или кадмием. Иногда развивается у работников химического производства, а именно – при контакте с толуолом, лизолом и малеиновой кислотой.
Иногда синдром Фанкони развивается при лечении препаратами платины, гентамицином и просроченными препаратами тетрациклиновой группы.
Симптомы синдрома Фанкони
Галерея некоторых симптомов
 Частое мочеиспускание
Частое мочеиспускание
 Искривление позвоночника
Искривление позвоночника
 Повышенная температура
Повышенная температура
У детей, как правило, основная часть симптомов связана с дефицитом фосфатов (похоже на фосфат-диабет). Появляется шаткая походка («утиная»), низкий рост, малоподвижность. Постепенно формируется О-образная форма голеней, искривляются другие кости скелета (особенно позвоночник). Из-за боли в костях ребёнок мало ходит. Очень высок риск переломов из-за костной ткани.
Дети растут необщительными и пугливыми – когнитивные (мыслительные) функции не страдают, однако формируются комплексы.

Запоры также могут быть связаны с болезнью Тони-Дебре-Фанкони
Первые признаки заболевания при врождённой форме появляются уже на первом году жизни (иногда – с 1,5 лет).
Ребёнок начинает часто мочиться, появляется (37-38 0С), развиваются запоры, может быть рвота.
Со временем, родители замечают вышеописанные симптомы дефицита фосфора. К 6 годам дети, как правило, уже не в состоянии самостоятельно ходить. При развитии процесса к 12 годам формируется , что чревато летальным исходом.
Нарушение обмена веществ приводит к патологиям нервной системы, проблемам со зрением, дефектами развития органов мочеполовой системы, заболеваниям кишечника и хроническому иммунодефициту.
У взрослых вторичный синдром проявляет себя (частое мочеиспускание), общей слабостью, мышечной гипотонией и болями в костях. Достаточно активно формируется почечная недостаточность, развивается артериальная гипертензия.
Хуже всего, когда приобретённый синдром Фанкони поражает женщин в постменопаузу. К естественному снижению плотности костной массы (остеопения, остеопороз) добавляется хрупкость костей из-за дефицита минералов. Заканчивается такая ситуация компрессионными переломами тел позвонков, переломами головки бедренной кости и инвалидизацией пациентки.

Остеопороз является вспомогательным симптомом при диагнозе синдрома Фанкони
Диагностика синдрома Фанкони
Синдром редкий, опытный врач может его заподозрить благодаря рентгену костей и некоторым расширенным биохимическим показателям крови и мочи.

Появление шпор связано с нарушением процесса формирования костной ткани
Синдром Фанкони нередко комбинируется с системным остеопорозом (остеопороз является вспомогательным симптомом, в данном случае). Костная ткань имеют грубоволокнистую структуру, очень часто видны костные выросты («шпоры»). Для определения плотности костной ткани используют денситометрию.
Кости слабо минерализованы, что определяется при анализе биоптата (образца ткани).
Со стороны почек – при исследовании определяется дистрофия почечных канальцев (по типу «лебединой шеи»), разрушается эпителий и происходит замещение почечных структур соединительной тканью.
Спасает больных только то, что клубочковый слой вовлекается в процесс последним, и до самого финала заболевания функция почек кое-как, но осуществляется.
При микроскопии эпителия клубочков в клетках видно большое количество митохондрий.
Лечение заболевания
Первичный синдром Фанкони излечению не подлежит, поскольку речь идёт о структурных изменениях в почках, а также стойких, генетически обусловленных метаболических нарушениях. Таким больным постоянно поддерживают уровень калия, лечат почечный канальцевый ацидоз и прочие дефекты водно-солевого обмена. Фосфат-диабет в этом случае подлежит стандартной для этого заболевания терапии.
Больные должны употреблять много жидкости в течении суток.
Галерея некоторых средств лечения синдрома Фанкони
 Капельницы
Капельницы
 Витамины
Витамины
 Хвойные ванны
Хвойные ванны
 Массаж
Массаж
Вторичный синдром Фанкони в ряде случаев излечим, особенно при успешном устранении заболевания или состояния, которое провоцирует эту почечную патологию.
Лекарственная терапия
Для компенсации нарушений обмена фосфора и кальция назначаются метаболиты витаминов группы Д – 1(ОН)D3 или 1,25(ОН)D3. Дозы витамина титруются от 10 000 МЕ до 100 000 МЕ в сутки. Дозировка подбирается под постоянным лабораторным контролям уровня фосфора и кальция в крови. Назначаются препараты кальция и фитин per os.
Такая терапия производится периодически, курсами.
Диетотерапия при синдроме Фанкони
Основной принцип диеты – ограничить выведение ряда веществ из организма, в т.ч. серосодержащих аминокислот и фосфора. Диета подразумевает ограничение соли и широкое использование ощелачивающих продуктов.
Необходимо ввести в рацион большое количество фруктовых соков и молока (при его нормальной переносимости)
 Фруктовые соки
Фруктовые соки
 Ограничение соли
Ограничение соли
 Молоко
Молоко
 Сухофрукты
Сухофрукты
Если нарушения опорно-двигательного аппарата становятся критическими, то рекомендуется хирургическая коррекция.
Своевременная диагностика нарушений обмена по типу синдрома Фанкони у детей позволяет избежать быстрой инвалидизации ребёнка, у взрослых – снизить риски переломов и ущемлений корешков нервных волокон, увеличить качество и продолжительность жизни. При появлении первых признаков заболевания (полиурия, изменение цвета мочи, боли в костях и суставах) обращайтесь к врачу.
Синдром де Тони - Дебре - Фанкони представляет собой тяжелое врожденное заболевание, характеризующееся разнообразными Страдают им чаще всего дети первого года жизни. Как правило, встречается в сочетании с другими наследственными патологиями, но может манифестировать и как самостоятельный синдром.
Краткий экскурс в историю
Заболевание было открыто и изучено в 1931 году доктором Фанкони из Швейцарии. Обследуя ребенка с рахитом, низким ростом и изменениями в анализах мочи, он пришел к выводу, что данное сочетание признаков стоит рассматривать как отдельную патологию. Спустя два года де Тони внес свои поправки, добавил к уже имеющемуся описанию гипофосфатемию, а еще через некоторое время Дебре выявил у подобных больных аминоацидурию.
В отечественной литературе это состояние называют терминами «наследственный синдром де Тони - Дебре - Фанкони» и «глюкоаминофосфатдиабет». За рубежом оно чаще именуется renal Fanconi syndrome.
Причины развития синдрома Фанкони
В настоящий момент так и не удалось до конца выяснить, что же лежит в основе этого тяжелого недуга. Синдром Фанкони предположительно является Специалисты считают, что развитие этой патологии связано с точечной мутацией, которая приводит к неправильной работе почек. Многочисленные исследования подтвердили, что в организме происходит нарушение клеточного обмена. Не исключено, что в деле замешан аденозинтрифосфат (АТФ) - соединение, играющее важную роль в обмене энергии. В результате некорректной работы ферментов теряется глюкоза, аминокислоты, фосфаты и другие не менее полезные вещества. В таких суровых условиях почечные канальцы не получают необходимой для их работы энергии. Полезные вещества выводятся вместе с мочой, нарушаются обменные процессы, развиваются рахитоподобные изменения костной ткани.
Синдром Фанкони у детей встречается гораздо чаще, чем у взрослых. По статистике частота патологии составляет 1:350 000 новорожденных. Болеют и мальчики, и девочки в равных пропорциях.
Признаки синдрома Фанкони
Заболевание может развиваться в любом возрасте, но чаще всего оно отмечается у детей первого года жизни. Глюкозурия, генерализованная гипераминоацидурия и гиперфосфатурия - эта триада признаков характеризует синдром Фанкони. Симптомы развиваются достаточно рано. Прежде всего родители замечают, что их ребенок начинает чаще мочиться, и его постоянно мучает жажда. Малыши, конечно же, не могут сказать об этом словами, но по их капризному поведению и постоянному зависанию на груди или бутылочке становится очевидно, что с ребенком что-то не так.

В дальнейшем родителям приносит много беспокойства частая беспричинная рвота, длительные запоры и не поддающаяся объяснению Как правило, на этом этапе ребенок наконец-то попадает на прием к врачу. Опытный педиатр может заподозрить, что на обычную простуду такое сочетание симптомов совсем не похоже. В случае если доктор окажется грамотным, он вовремя сможет распознать синдром Фанкони.
Симптомы между тем никуда не исчезают. К ним добавляется заметное отставание в физическом и умственном развитии, появляются выраженные искривления крупных костей. Обычно изменения затрагивают лишь нижние конечности, приводя к деформации по варусному или вальгусному типу. В первом случае ножки ребенка будут искривлены колесом, во втором - в виде буквы «Х». И тот и другой вариант, конечно же, неблагоприятны для дальнейшей жизни ребенка.
Синдром Фанкони у детей часто включает в себя остеопороз (преждевременное разрушение костной ткани), а также значительную задержку роста. Не исключены переломы длинных и параличи. Даже если до сих пор родители не беспокоились о состоянии малыша, то на этом этапе они уже точно не откажутся от квалифицированной помощи.

Синдром Фанкони у взрослых встречается довольно редко. Все дело в том, что это серьезное заболевание закономерно приводит к развитию почечной недостаточности. При таком раскладе невозможно дать какой-либо однозначный прогноз и гарантировать большую В литературе описаны случаи, когда в возрасте 7-8 лет синдром Фанкони сдавал свои позиции, наступало заметное улучшение состояния ребенка и даже выздоровление. К сожалению, такие варианты в современной практике достаточно редки, чтобы можно было сделать какие-то серьезные выводы.
Диагностика синдрома Фанкони
Кроме сбора анамнеза и тщательного осмотра доктор обязательно назначит некоторые обследования, позволяющие подтвердить данное заболевание. Синдром Фанкони неизбежно приводит к нарушению работы почек, а значит обязательным будет обычный анализ мочи. Конечно же, этого недостаточно, чтобы выявить все особенности течения болезни. Необходимо посмотреть не только содержание в моче белка и лейкоцитов, но и попытаться обнаружить лизоцим, иммуноглобулины и другие вещества. В анализе также обязательно выявится высокое содержание сахара (глюкозурия), фосфатов (фосфатурия), будут видны значительные потери важных для организма веществ. Подобное обследование можно провести как амбулаторно, так и в условиях стационара.

В анализах крови тоже неизбежны некоторые изменения. При биохимическом исследовании отмечается снижение почти всех значимых микроэлементов (прежде всего кальция и фосфора). Развивается выраженный мешающий нормальной работе всего организма.
Рентгенография скелета покажет остеопороз (разрушение костной ткани) и деформацию конечностей. В большинстве случаев обнаруживается отставание темпов роста костей и их несоответствие биологическому возрасту. При необходимости доктор может назначить УЗИ почек и других внутренних органов, а также обследование у смежных специалистов.
Дифференциальный диагноз
Бывают случаи, когда некоторые другие заболевания маскируются под синдром Фанкони. Перед врачом встает сложная задача разобраться, что же происходит с маленьким пациентом на самом деле. Иногда глюкоаминофосфатдиабет путают с хроническим пиелонефритом и другими заболеваниями почек. Изменения в анализах мочи, а также характерные особенности поражения костной ткани помогут педиатру выставить верный диагноз.
Лечение синдрома Фанкони
Стоит учесть тот факт, что данная патология является хронической. Довольно сложно полностью избавиться от неприятных симптомов, можно лишь на какое-то время уменьшить проявления болезни. Что же предлагает современная медицина в качестве помощи больным детям?
На первом месте стоит диета. Пациентам рекомендуют ограничить употребление соли, а также всех острых и копченых продуктов. В рацион добавляют молоко и различные фруктовые сладкие соки. Не стоит забывать и о (чернослив, курага и изюм). В том случае, когда дефицит микроэлементов дошел до критической стадии, врачи назначают прием специальных витаминных комплексов.
На фоне диеты вводят большие дозы витамина D. Состояние пациента постоянно контролируется - ему приходится время от времени сдавать кровь и мочу на анализы. Это необходимо для того, чтобы вовремя выявить начинающийся гипервитаминоз и снизить дозу витамина D. Лечение длительное, большими курсами, с перерывами. В большинстве случаев такая терапия помогает восстановить нарушенный обмен веществ и предотвратить тяжелые осложнения.

Если болезнь зашла далеко, пациент попадает в руки хирургов. Опытные ортопеды сумеют выправить костные деформации и существенно повысить уровень жизни ребенка. Подобные операции выполняются только в случае стойкой и длительной ремиссии: не менее полутора лет.
Прогноз
К сожалению, прогноз у таких пациентов неблагоприятный. В большинстве случаев болезнь медленно прогрессирует, рано или поздно приводя к почечной недостаточности. Деформации костей скелета неизбежно ведут к инвалидизации и ухудшению общего качества жизни.
Можно ли избежать этой патологии? Несомненно, подобный вопрос волнует каждого, кто столкнулся с синдромом Фанкони. Родители пытаются понять, что они сделали не так и где не уследили за ребенком. Не менее важно знать, грозит ли повторение ситуации с другими детьми. К сожалению, меры профилактики в настоящий момент не разработаны. Парам, планирующим обзавестись еще одним ребенком, стоит проконсультироваться у генетика для получения более полной информации о волнующей их проблеме.
Синдром Висслера - Фанкони (аллергический субсепсис)
Описано это заболевание только у детей от 4 до 12 лет. Причина этой серьезной патологии до сих пор не известна. Можно предположить, что этот синдром является типичным аутоиммунным заболеванием, особой формой ревматоидного артрита. Начинается всегда остро, с подъема температуры, которая неделями может держаться на цифре 39 градусов. Во всех случаях появляется полиморфная сыпь на конечностях, иногда на лице, груди или животе. Обычно происходит выздоровление без каких-либо серьезных осложнений. Однако же у части маленьких пациентов со временем развиваются тяжелые поражения суставов, приводящие к инвалидизации.
Добавляю еще информации по данному синдрому.
=====================
Болезнь Дебре — Де Тони — Фанкони
Болезнь Дебре-де Тони-Фанкоии — наследственное заболевание, передающееся по аутосомно-рецессивному типу. В основе патогенеза его лежит нарушение канальциевого транспорта аминокислот, глюкозы, неорганических фосфатов и гидрокарбонатов. Снижена канальциевая реабсорбция натрия, калия, воды, что обусловлено генетически детерминированной дисплазией нефронов. Тяжесть состояния определяется поражением не только тубулярного аппарата, но и клубочков почек. Нарушение реабсорбции воды сопровождается полиурией, полидипсией, гипостенурией и рецидивами лихорадки без видимых причин. Заболевание обычно начинается на первом году жизни. На втором году жизни у ребенка выявляются отставание в физическом развитии и костные деформации грудной клетки, костей предплечий, плеча, голеней, таза.
При рентгенографии костей обнаруживаются выраженные деформации конечностей, нарушение структуры костной ткани (системный остеопороз различной степени выраженности, истончение коркового слоя трубчатых костей, разрыхление зон роста, отставание темпов роста костной ткани от календарного возраста ребенка). Характерными особенностями болезни Дебре- де Тони -Фанкони являются понижение концентрации кальция в крови ниже 2,21 ммоль/л и фосфора до 0,9 ммоль/л, повышение активности щелочной фосфатазы в крови, метаболический ацидоз. Экскреция кальция с мочой остается нормальной (1,5-3,5 ммоль/сут), а клиренс фосфора мочи обычно повышен. Одновременно выявляется глюкозурия (2-3% и выше), генерализованная аминоацидурия (до 2-2,5 г/сут), полиурия - до 2 л/сут и более, повышение рН мочи до 6,0. Относительная плотность мочи может быть высокой (1,025-1,035). При экскреторной урографии могут находить изменения контуров почек, повышение их подвижности, наличие аномалии сосудов.
Выделяют два варианта болезни Дебре-де Тони-Фанкони.
1-й — характеризуется грубой задержкой физического развития (дефицит длины тела более 20%), тяжелым течением заболевания, выраженными деформациями костей и нередко переломами костей, выраженной гипокальциемией (1,6-1,8 ммоль/л), снижением всасывания кальция в кишечнике (менее 20%).
2-й — характеризуется умеренной задержкой физического развития (дефицит длины тела менее 13%), легким течением заболевания, умеренными деформациями костей, нормальной концентрацией кальция в крови.
Лечение. Для ликвидации нарушений фосфорно-калыщевого обмена применяется витамин D. Начинают лечение с 10 000 ME, постепенно повышая дозу до 25 000-30 000 ME, а затем до 75 000- 150 000 ME. Повышение начальных доз проводится осторожно, постепенно каждые 2 нед, до нормализации концентрации кальция и фосфора в крови. Из активных метаболитов витамина D можно использовать оксидевит в дозе 0,5-1,5 мкг/сут. В комплекс лечения включают также препараты кальция (кальция глюконат, 1,5-2 г/сут) и фосфора (0,5-1 г/сут). Лечение витамином D должно проводиться повторными курсами, так как при отмене препарата часто наступают кризы метаболического прогрессирования остеопороза и рахитических изменений в костной систем. Диета должна быть с ограничением поваренной соли и включением пищевых продуктов, оказывающих ощелачивающее действие (молоко, молочные продукты), фруктовых соков, богатых калием сухофруктов (чернослив, курага, изюм и др.), витаминов А, В, С, Е. Показаны также соляно-хвойные ванны.
=========================
Синдром Фанкони (де Тони-Дебре-Фанкони) рассматривают как «большую» канальцевую дисфункцию, характеризующуюся нарушением реабсорбции большинства веществ и ионов (аминоацидурией, глюкозурией, гиперфосфатурией, увеличением экскреции бикарбоната) и системными метаболическими изменениями.
Синдром Фанкони включает множественные дефекты реабсорбции в проксимальных почечных канальцах, что ведет к глюкозурии, фосфатурии, генерализованной аминоацидурии и уменьшению концентрации гидрокарбонатов. Симптомы у детей включают гипотрофию, задержку физического развития и рахит, симптомы у взрослых — остеомаляцию и мышечную слабость. Диагноз основывается на выявлении глюкозурии, фосфатурии и аминоацидурии. Лечение включает возмещение дефицита гидрокарбонатов, а также лечение почечной недостаточности.
| Синдром де Тони - Дебре - Фанкони | |
|---|---|
| МКБ-10 | 72.0 72.0 |
| МКБ-9 | 270.0 270.0 |
| OMIM | , , , , и |
| DiseasesDB | |
| MedlinePlus | |
| eMedicine | ped/756 |
| MeSH | D005198 |
История
Тип наследования - аутосомно-рециссивный, выделена также аутосомно-доминантная форма с локализацией гена на хромосоме 15q15.3. Экспрессивность мутантного гена в гомозиготном состоянии значительно варьирует. Встречаются спорадические случаи, обусловленные свежей мутацией. Считается, что в основе болезни лежат генетически обусловленные дефекты ферментативного фосфорилирования в почечных канальцах (комбинированная тубулопатия), дефицит ферментов 2-го и 3-го комплексов дыхательной цепи - сукцинатдегидрогеназного и цитохромоксидазного. Учёные относят заболевание к разряду митохондриальных болезней [ ] .
Патогенез
Патологические изменения представляют собой один из вариантов вторичного гиперпаратиреоза . Основное звено патогенеза - митохондриальный ферментный дефект в цикле Кребса , ферментная тубулопатия , характеризующаяся нарушением реабсорбции глюкозы , аминокислот , фосфатов и бикарбонатов в канальцах почек . Потеря аминокислот и бикарбоната способствует развитию метаболического ацидоза, на фоне которого усиливается резорбция костной ткани и снижается реабсорбция калия и кальция в канальцах почек, что приводит к развитию гипокалиемии и гиперкальциурии. Потеря фосфора ведёт к развитию рахита, а у детей старшего возраста и взрослых - к остеомаляции .
Таким образом, митохондриальный ферментный дефект в цикле Кребса ведёт к нарушению процессов энергообеспечения реабсорбции фосфатов, глюкозы и аминокислот в почечных канальцах и повышенной их экскреции с мочой - нарушается кислотно-основное равновесие , а метаболический ацидоз и недостаток фосфатов способствуют разрушению костной ткани по типу рахитоподобных изменений скелета и остеомаляции.
Клиническая картина
Первые признаки заболевания появляются во втором полугодии жизни - дети вялые, гипотрофичные , аппетит резко снижен, наблюдаются рвота , субфебрилитет , гипотония , жажда , полиурия , дегидратация . Развёрнутый симптомокомплекс формируется ко второму году жизни. Если заболевание манифестирует в 5-6 лет, то первыми признаками являются симптомы остеомаляции , деформация костей и гипокалиемические параличи . Со второго года жизни выявляют отставание физического и интеллектуального развития, происходит генерализованная декальцификация , проявляющаяся костными деформациями ног (вальгусные или варусные), грудной клетки, предплечий и плечевых костей, снижение мышечного тонуса. Рентгенологически выявляют деформации костей, позвоночного столба, переломы , системный остеопороз различной степени выраженности, истончение коркового слоя трубчатых костей, разрыхление зон роста, отставание темпов роста костной ткани от паспортного возраста ребёнка. Кости становятся ломкими.
При лабораторном обследовании выявляют нормо- или гипокальциемию , гипофосфатемию, повышенный уровень щелочной фосфатазы . В результате снижения реабсорбции бикарбонатов в канальцах почек наблюдается гиперхлоремический ацидоз на фоне избытка паратгормона и нормо- или гипокальциемии. В биохимическом анализе мочи обнаруживают аминоацидурию, глюкозурию (при нормальных уровнях гликемии), натрийурию, гипокальцийурию на фоне гиперфосфатурии .
В зависимости от тяжести клинических проявлений и метаболических расстройств выделяют два клинико-биохимических варианта болезни:
- Первый характеризуется значительной задержкой физического развития, тяжёлым течением заболевания с выраженными костными деформациями и нередко переломами костей, резкой гипокальциемией (1,6-1,8 ммоль/л), снижением абсорбции кальция в кишечнике.
- При втором варианте отмечают умеренную задержку физического развития, лёгкое течение с незначительными костными деформациями, нормокальциемию и нормальное усвоение кальция в кишечнике.
Биохимические нарушения
- снижение уровня кальция в крови;
- снижение уровня фосфора в крови;
- повышение уровня щелочной фосфатазы;
- развитие метаболического ацидоза (рН : 7,35…7,25; ВЕ: −10…-12 ммоль/л) за счёт дефекта реабсорбции бикарбонатов в проксимальных канальцах;
- нормальная экскреция кальция с мочой;
- повышение клиренса фосфатов мочи, всасывание фосфатов в кишечнике не страдает;
- развитие глюкозурии (20-30 г/л и выше);
- развитие генерализованной гипераминоацидурии;
- нарушение функций аммониоацидогенеза - снижение титрационной кислотности, повышение рН мочи больше 6,0;
- развитие гипокалиемии .
Исходом заболевания является развитие хронической почечной недостаточности .
Дифференциальный диагноз
Дифференциальную диагностику синдрома де Тони-Дебре-Фанкони проводят с
Синдром Фанкони (полное название – де Тони-Дебре-Фанкони) – врожденная патология, которая выражается в серьезной дисфункции проксимальных почечных канальцев, а именно – нарушении вторичной абсорбции (всасывании в кровь) отфильтрованных почками веществ, которое приводит к глюкозурии (повышенному сахару в моче), фосфатурии (нарушению обмена фосфора и кальция), аминоацидурии (повышенному выведению аминокислот с мочой) и уменьшению концентрации гидрокарбонатов, регулирующих кислотность крови.
Синдром Де-Тони-Дебре-Фанкони
Синдром Фанкони очень редкое заболевание, в основном встречающаяся у детей, и по медицинской статистике ее частота соответствует 1 больному малышу на 350 тысяч новорожденных обоих полов.
У взрослых людей наблюдается крайне редко, развиваясь на фоне приобретенных патологий. Код патологии согласно МКБ-10: E72.О.
Причины
Характер и причины генетического порока синдрома Фанкони сегодня изучены недостаточно.
Предположено, что в основе патологии лежат или пороки транспортных белков почечных канальцев или генная мутация, искажающая функцию ферментов, регулирующих обратное всасывание глюкозы, аминокислот и фосфора.
Имеются данные исследований о точечных дефектах митохондрий, приводящих к неправильному функционированию канальцев почек.
Болезнь также связывают с непереносимостью фруктозы, хроническим отравлением токсинами (тяжелых металлов, ифосфамида, аминогликозидов), дефицитом витамина D, амилоидозом, недостаточностью целого ряда клеточных ферментов (пируваткарбоксилазы, фосфоенолпируваткарбоксикиназы и прочих), тирозинемией, метахроматической лейкодистрофией, галактоземией, цистинозом, гликогенозами.
По мнению других специалистов, синдром Фанкони может являться изолированной патологией – а именно – одной из тяжелых форм рахитоподобных патологий, имеющих наследственный характер.
Исследования подтверждают, что при синдроме Фанкони нарушен клеточный энергетический обмен с участием АТФ (аденозинтрифосфат) и межклеточный транспорт в канальцах главного элемента почки - нефрона.
Вследствие недостаточности функций ферментов происходят потери глюкозы, фосфатов, аминокислот, и канальцы почек испытывают дефицит энергии. При этом важные вещества уходят с мочой, приводя к дистрофическим изменениям костной ткани – рахиту.
Так как в медицине пока не пришли к однозначному выводу о причинах синдрома Факони, это состояние обозначают и другими терминами: «глюкофосфаминный диабет», «идиопатический ренальный синдром Фанкони», «D-резистентный рахит», «почечный нанизм с D-резистентным рахитом», «наследственный синдром Фанкони».
Формы и патогенез
Выделяют два вида синдрома Фанкони:
- наследственный (врожденный, идиопатический), который относится к первичной форме;
- приобретенный, который рассматривают как вторичную форму болезни.
Наследственный (генетический) вид специалисты связывают с дефектом X-хромосомы, который наследуется по доминантному и рецессивному типу, поэтому генетический прогноз его проявления у будущего потомства – задача не простая. Если патология относится к врожденному (первичная форма) типу, то она выявляется у ребенка в грудном периоде до года. Поэтому первичную форму называют «младенческой».
Степень генной мутации обуславливает и степень выраженности самого синдрома. Так, наследственный полный синдром Фанкони обнаруживает себя при наличии 3 базовых биохимических дефектов, к которым относят глюкозурию, аминоацидурию, фосфатурию, неполный - при двух из них.
Обычно генетически обусловленная патология сопровождается другими врожденными недугами: цистинозом, синдромами Вильсона, Дента, Лоу, непереносимостью фруктозы, тирозинемией (неспособность организма эффективно расщеплять аминокислоту тирозин), галактоземией (дисфункция преобразования сахара в глюкозу), избыточной аккумуляцией гликогена.
Развитие синдрома Фанкони
Приобретенный синдром (вторичный), в отличие от врожденного, не сопровождается, а является следствием уже имеющихся патологий:
- тирозинемия I типа;
- цистиноз (нарушение обмена аминокислоты цистина с последующим поражением почек);
- непереносимость фруктозы;
- болезнь Вильсона-Коновалова;
- галактоземия;
- гликогеноз (аномальное скопление гликогена в тканях и органах) тип XI;
- наследственные патологии почек;
- (нарушение белкового обмена, приводящее к склерозу, атрофии, дисфункции органов);
- тубулоинтерстициальные (нефриты с поражением ткани, канальцев почек);
- гиперпаратиреоз (эндокринное заболевание, при котором нарушается нормальное содержание в крови кальция и фосфора);
- злокачественные образования: миелома, легких, поджелудочной железы, легких, болезнь легких цепей, ;
- глубокие ожоги.
Кроме того, синдром могут спровоцировать такие состояния:
- пересадка органа при низкой совместимости тканей;
- дефицит витамина D;
- отравление ураном, висмутом, ртутью, свинцом, кадмием;
- контакт с толуолом, малеиновой кислотой, лизолом;
- применение нефротоксичных фармакологических средств, таких как: Гентамицин, препараты платины, просроченные лекарства на основе тетрациклина, Диданозин, Цидофовир, медикаменты противораковой химиотерапии – Ифосфамид, Стрептозоцин.
Симптомы и признаки
При наследственной (врожденной) форме
Первичные симптомы проявляются в первые месяцы жизни, редко – после полутора лет.
Прежде всего, у новорожденного ребенка замечают следующие состояния:
- частое мочеиспускание (полиурия);
- повышенная жажда (полидипсия);
- длительные запоры;
- частые приступы беспричинной рвоты;
- астения (общая утомляемость), мышечная слабость;
- необъяснимые «скачки» температуры до 37,5 – 38 C;
- вздутый животик.
Как правило, в период, когда начинаются приступы рвоты и подъемы температуры малыша показывают педиатру. Опытный специалист должен определить, что сочетание беспокоящих родителей признаков не имеет отношения к ОРЗ, ОРВИ или энтеровирусной инфекции.
Грамотный детский врач способен вовремя распознать синдром Фанкони. А лабораторные исследования – подтвердить подозрения, если выявляют три (или два) базовых признака: глюкозурию, генерализованную гипераминоацидурию и гиперфосфатурию, характерных для этой патологии.
После слабовыраженных и достаточно неопределенных симптомов в последующие год – полтора четко фиксируются свойственные синдрому Фанкони симптомы:
- Ранний нанизм (низкорослость), вызванный постоянным выведением из организма важнейших аминокислот, глюкозы, кальция, фосфатов. Первые полгода нормального роста и веса сменяются дефицитом массы тела (до 30%) и роста (от 2 до 21%).
- Рахит, обусловленный массивным выведением кальция и фосфатов, становится заметным после 10 – 12 месяцев жизни, причем имеет характерные для синдрома Фанкони особенности: головка малыша обычно мало деформирована, но крупные кости ножек и ручек показывают существенные искривления - деформации по варусному типу, когда голени малыша искривляются «колесом», или вальгусному (в виде буквы «Х»). Искривляются и кости грудной клетки, позвоночника.
- Задержка в умственном и физическом развитии.
- Необщительность, пугливость, закомплексованность.
- Полидипсия и полиурия могут прогрессировать и регрессировать, не проходя окончательно.
- Умеренная мышечная гипотония, выражающаяся в замедленности, затрудненности движений, приводящая к тому, что дети 5 – 6 лет не способны ходить.
- Боли в костях, умеренной интенсивности, мешающие ребенку ходить. Сильнее проявляются на уровне ног, таза и позвоночного столба. Походка, если ребенок ходит, становится «утиной», неуверенной.
- Высокая вероятность переломов трубчатых костей из-за дефицита минералов в костной ткани.
- Остеомаляция или размягчение костей из-за разрушения костной ткани в результате нехватки солей кальция и фосфора.
- Сниженная иммунная защита к инфекциям, что проявляется в частых вирусных заболеваниях, отитах, пневмониях.
- Параличи, вызванные нехваткой калия.
- Офтальмологические патологии, такие как: пигментный ретинит, врожденная катаракта.
- Развитие патологий нервной системы, ЛОР (ухо, нос, глотка, гортань) и ЖКТ-органов, сердечно-сосудистой системы, анатомические аномалии органов мочевой системы вследствие массивных нарушений обмена веществ.
- В единичных случаях - эндокринные нарушения
При прогрессировании тубулярных расстройств (нарушение транспорта органических веществ, минералов, электролитов) к 10 – 12 годам у детей повышена вероятность развития хронической почечной недостаточности, что угрожает его жизни.
Видимые симптомы синдрома Фанкони у детей

У взрослых пациентов при развитии вторичной формы
Если приобретенный синдром Фанкони развивается у людей взрослых при других болезнях или патологических состояниях, то его проявления, часто сочетаются с проявлениями заболевания-провокатора.
Тем не менее, выявлены базовые признаки:
- Увеличенный объем мочи в сутки (до 2 литров и более) и острая жажда, свойственные также пациентам раннего детского возраста.
- Общая и мышечная слабость, боли в костях.
- Высокая вероятность упорного повышения кровяного давления () на фоне дисфункции почек.
- Остеомаляция (разрушение костей).
- Ацидоз (повышение кислотности крови) по причине задержки продуктов окисления в организме, приводящий к гипокалиемии (дефициту калия).
- Нефрокальциноз - высокое отложение солей кальция в почках с характерными для этого состояния лихорадкой, ознобом, тошнотой, сильнейшими болями в животе, паху, яичниках.
- Гипокалиемия (низкое поступление калия), вызывающая тяжелые сердечные осложнения, включая угрожающие жизни аритмии.
- Быстрое (при отсутствии лечения) формирование хронической недостаточности почек
У женщин
Наиболее неблагоприятный вариант течения синдрома Фанкони реализуется при его развитии у женщин в периоде постменопаузы. В это время, на фоне снижения продукции гормонов происходит естественное уменьшение плотности костной ткани (остеопения).
При сочетании этого состояния с увеличением хрупкости костей по причине нехватки минералов, высока вероятность тяжелых компрессионных переломов позвонков, шейки бедра и последующей инвалидностью.
Диагностика
Чтобы подвердить или опровергнуть диагноз, с помощью рентегографии проводят обследование костей и углубленные биохимические исследования крови и мочи.
Лабораторная
Выявляемые изменения в биохимии мочи и крови:
| Признаки | Показатели |
|---|---|
| низкое содержание кальция и фосфора | менее 2,1 ммоль/л и 0,9 ммоль/л соответственно |
| ацидоз («закисление» крови) | ВЕ = 10 – 12 ммоль/л |
| глюкозурия (увеличение сахара в моче) | 2 – 3% и выше |
| гипераминоцидурия (выведение с мочой важных аминокислот аланина, аргинина, глицина, пролита) | до 2 – 2,5 г/сутки |
| выведение кальция с мочой | 1,5 – 3,5 ммоль/сут |
| увеличение рН (кислотности) мочи из-за аномально высокой потери бикарбонатов | до 6,0 |
| увеличение относительной плотности мочи | 1,025 – 1,035 |
При обследовании также обнаруживают:
- увеличение активности щелочной фосфатазы;
- чрезмерное выведение из организма солей натрия, калия;
- протеинурию (появление белка в моче) при наличии легких цепей иммуноглобулинов, лизоцима, низкомолекулярных белков, бета 2-микроглобулинов;
- повышение клиренса (скорости фильтрации) мочевой кислоты с ее пониженным сывороточным содержанием
- падение активности ферментов энергообмена: сукцинатдегидрогеназы, а-глицерофосфатдегидрогеназы, глутаматдегидрогеназы;
- повышение в крови количества молочной и пировиноградной кислоты.
Инструментальная
Диагностирование синдрома Фанкони предусматривает обязательное использование рентгенографии костей с целью выявления деформации скелета, конечностей, обнаружения признаков остеомаляции, остеопороза, а у детей – дополнительно – задержки роста костей в сравнении с нормой по календарному возрасту.
Наблюдаются следующие аномалии в костной ткани:
- грубоволокнистая, ячеистая структура с лакунами, слабой минерализацией, аномальных разрастаний в виде «шипов» в бедренных и берцовых костях;
- признаки эпифизиолиза (частичная или полная остановка роста кости в длину в несозревшем скелете, приводящее к асимметрии конечностей);
- переломы трубчатых костей (на поздней стадии) на фоне развития остеопороза, степень которого определяют с помощью рентгеноденситометрии;
- аккумуляция радиоизотопа в областях интенсивного роста кости.
В почках:
При электронном исследовании биопсии ткани почки (биопсии) выявляют характерное изменение формы канальцев в виде «шеи лебедя», истончение, атрофию (уменьшение объема) эпителиальной ткани при наличии повышенного количества митохондрий в ней, фиброз (аномальное разрастание) соединительной ткани.
Необходимые исследования:
Дифференциальная
Патологию следует отличать от всех изолированно протекающих болезней, которые способны провоцировать возникновение приобретенного синдрома Фанкони, приобретенных нездоровых состояний и интоксикаций.
Кроме того, у грудничков педиатр обязан дифференцировать состояние острой нехватки витамина D при синдроме Фанкони от его переизбытка при употреблении искусственных добавок или расстройстве кальциевого обмена.
Различия при гипервитаминозе D и синдроме де Фанкони у детей до года:
| Параметры | Гипервитаминоз D | Синдром де Фанкони |
|---|---|---|
| Частота | Часто | Редко |
| Симптомы (схожие) | Сухость кожи, бледность, острая жажда, рвота, длительные запоры, дефицит веса и роста, увеличение печени | |
| Симптомы (различающиеся) | Гипертензия (частые подъемы кровяного давления) | Истощение, полиурия, гипотония мышц, повышенное давление крови отсутствует |
| Кровь | Избыток кальция в остром периоде. Уменьшенное содержание фосфора. Щелочная фосфатаза, сахар, белок - в норме | Кальций чаще в норме (может быть понижен). Глюкоза, белок снижены. Фосфор снижен резко. Активность щелочной фосфатазы резко повышена - в 2 – 3 раза. Кислотность крови повышена |
| Моча | Тест Сулковича на выведение кальция положительный. Наличие белка, крови (более 2 – 3 эритроцитов в поле зрения), лейкоцитов. Сахар аминоазот, как правило, в норме | Тест Сулковича отрицательный. Белок, фосфаты, сахар повышены, аминоацидурия |
| Кости | Зоны обызвествления расширены, уплотнены | Остеопороз трубчатых костей, дефицит кальция в областях обызвествления |
Специалисты, занимающиеся синдромом Фанкони и его осложнениями: нефролог, ортопед, гематолог, эндокринолог, уролог, офтальмолог, генетик.
Лечение
Рациональная, хорошо продуманная терапия способна уменьшить влияние на мозг, костную систему и органы чрезмерных потерь важнейших аминокислот, минералов, глюкозы и белка, выводимых в составе мочи.
Поэтому лечение при синдроме нацелено на следующие задачи:
- Максимально возможная коррекция дефицита калия, бикарбонатов, изменений в кислотно-щелочном балансе крови для уменьшения ацидоза.
- Терапия фосфат-диабета (D-резистентного рахита) с акцентом на недопустимости ограничения жидкостей.
- Лечение основного заболевания, провоцирующего развитие приобретенного синдрома у взрослых.
Медикаментозное
 Для смягчения потерь фосфора и кальция используют специальные препараты с витамином D, это l,25(OH)D3 и l(OH)D3.
Для смягчения потерь фосфора и кальция используют специальные препараты с витамином D, это l,25(OH)D3 и l(OH)D3.
Начальные дозы витамина D3 в сутки 10 – 15 тыс. ME. Повышение дозы проводят постепенно, увеличивая ее каждые 12 – 14 дней (под контролем пробы Сулковича и содержания в крови фосфора). При отсутствии признаков интоксикации и небольшом выведении кальция с мочой разрешается увеличивать дозу, доводя до 100 – 150 тыс. ME в сутки, и продолжать терапию до нормальных показателей в крови фосфора и щелочной фосфатазы. При стабилизации их значений повышать дозу далее не следует.
Терапия витамином D проводится несколькими курсами с целью предупреждения кризов прогрессирования рахитических деформаций в костях.
Оптимальным вариантом считают использование активных метаболитов D3 – Оксидевит (0,5 – 1,5 мкг в день), кальциотриол (Рокальтрол).
Включают препараты кальция (кальция глюконат в сутки до 1,5 – 2 грамм), фосфора (0,5 – 1 грамма в сутки), фитина.
Из неорганических фосфатов используют смесь Олбрайта, принимая его по 1 большой ложке 4 – 5 раз в сутки. Применяют фосфаты в виде раствора и таблеток в дозах, рассчитанных по 10 мг на килограмм веса, 4 раза в день (обязательно с препаратами витамина D во избежание гиперпаратиреоза).
При выраженном дефиците калия применяют Панангин, Аспаркам.
При любых назначениях постоянно отслеживают КОС или кислотно-основное состояние крови. В норме кровь имеет слабощелочную реакцию, а рН в диапазоне 7,35 – 7,45. При ацидозе, когда величина рН снижается ниже 7,35, кровь приобретает повышенную кислотность.
В этих случаях показано внутривенное вливание раствора натрия гидрокарбоната 4% или питье раствора (50 – 60 мл в день), в который входит лимонная кислота — 2 грамма, цитрат натрия – 3 г, цитрат калия – 3,3 г, вода 100 мл. В 1 мл такой ощелачивающей микстуры содержится по 1 ммоль натрия и калия. Высокая кислотность крови нейтрализуется также с помощью питьевой соды (бикарбоната натрия).
Некоторые специалисты на основе практических результатов рекомендуют Унитиол как средство, повышающее активность тиолзависимых ферментов.
При цистинозе назначают: Дитиотрентал из расчета 25 мг на килограмм веса пациента через 3 часа; Цистеамин в суточной дозе из расчета 90 мг/кг.
Имеются положительные результаты применения Пеницилламина, понижающего в крови концентрацию пировиноградной кислоты, уменьшающего степень выведения аминокислот и способствующего росту в организме резервов щелочи.
Хорошее влияние оказывают на работу канальцев почек гормональные анаболики, включая Метилтестостерон.
Лечение в стационаре показано при явно выраженных обменных нарушениях, включая гипо- и гипергликемию, деформациях скелета.
Профилактика
Современная профилактика врожденного синдрома Фанкони при наличии подобной патологии в семье заключается в предварительном генетическом консультировании. Риск развития болезни для так называемых сибсов, то есть, сестер и братьев - около 25%.
При вторичной форме синдрома его симптомы уменьшаются или полностью исчезают при активном лечении основной болезни или приобретенного патологического состояния.
Прогноз
Прогноз заболевания зависит от формы (первичная, вторичная) синдрома, тяжести проявлений, и начала лечения.
Например, симптоматика приобретенного синдрома исчезает при устранении причины-провокатора.
При врожденной патологии на фоне отсутствия отложений цистина в тканях течение болезни не несет серьезной угрозы жизни, в ином случае, особенно, без соответствующего лечения, гибель пациента прогнозируется до 10 – 20 лет от нарастающей недостаточности почек.
Однако, даже при тяжелых изменениях в почках: пиелонефрите, тубулоинтерстициальном нефрите, почечной недостаточности, при раннем медикаментозном воздействии на патологический процесс и длительной терапии устанавливается определенное равновесие в гомеостазе, при котором прогноз нормального качества жизни на десятки лет – хороший.
В медицинской практике встречаются истории болезни, когда у детей 7 – 8 лет наследственный синдром Фанкони практически «купировался» с наступлением длительной ремиссии, явного улучшения состояния ребенка и даже – выздоровления.