Amiotrofia muscular espinal tipo 1. Tipos de sma. Síntomas y tratamiento de la artrosis de la columna cervical.
La atrofia muscular espinal se manifiesta en la primera infancia. Los primeros síntomas pueden aparecer a los 2-4 meses de edad. Esta es una enfermedad hereditaria, que se caracteriza por el hecho de que en el tronco del encéfalo mueren gradualmente. células nerviosas.
tipos de problemas
Dependiendo de cuándo aparece por primera vez, la gravedad del curso y la naturaleza de los cambios atróficos, se distinguen varios tipos de enfermedad.
La atrofia muscular espinal puede desarrollarse debido a:
Primer tipo: agudo (forma de Werdig-Hoffman);
El segundo tipo: intermedio (infantil, crónico);
Tercer tipo: forma de Kugelberg-Welander (crónica, juvenil).
Tres tipos de una misma enfermedad surgen, según los expertos, debido a diferentes mutaciones el mismo gen. La atrofia muscular espinal es una enfermedad autoinmune que ocurre cuando se heredan dos genes recesivos, uno de cada padre. El sitio de mutación se encuentra en el cromosoma 5. Está presente en cada 40 personas. El gen se encarga de codificar una proteína que asegura la existencia de neuronas motoras en la médula espinal. Si este proceso se interrumpe, las neuronas mueren.
Enfermedad de Verdin-Hoffman
Puedes sospechar la presencia de un problema incluso durante el embarazo. Si un niño desarrolla atrofia muscular espinal tipo 1, a menudo se observan movimientos fetales lentos y tardíos durante el embarazo. Después del nacimiento, los médicos pueden diagnosticar hipotonía muscular generalizada.

Las atrofias comienzan a aparecer en los primeros meses de vida. En las partes proximales a menudo se observan lesiones fasciculares de la espalda, el tronco y las extremidades. También se observan trastornos bulbares. Estos incluyen succión lenta, llanto débil y dificultad para tragar. Los niños con enfermedad de Verdin-Hoffman a menudo experimentan una disminución de los reflejos nauseoso, tos y faríngeo y palatino. También experimentan fibrilación de los músculos de la lengua.
Pero estos no son todos los signos por los que se puede determinar la atrofia muscular espinal. Los síntomas característicos del tipo 1 de esta enfermedad incluyen debilidad de los músculos intercostales. Al mismo tiempo, el pecho de los bebés parece aplanado.
En los primeros meses de vida, estos niños suelen sufrir infecciones respiratorias, neumonía y aspiraciones frecuentes.
Diagnóstico de la enfermedad de Verdin-Hoffman
La enfermedad se puede determinar examinando al niño. Existen una serie de signos clínicos mediante los cuales los especialistas pueden determinar que un bebé padece atrofia muscular espinal hereditaria. Los diagnósticos incluyen:
Análisis de sangre bioquímico (mostrará un ligero aumento de aldolasa y creatina fosfoquinasa);
Estudio electromiográfico (el ritmo en empalizada indicará daño en los cuernos anteriores de la médula espinal);
Frecuencia de enfermedades
La AME es una de las enfermedades huérfanas (raras) más comunes y afecta a uno de cada 6.000-10.000 recién nacidos.
Causa de la AME
La AME es una enfermedad hereditaria y está asociada con mutaciones en el gen SMN1.
Para que la enfermedad se manifieste, ambos padres deben ser portadores de una mutación en este gen. Aproximadamente una de cada 40 personas tiene el gen recesivo AME. La probabilidad de tener un hijo enfermo de dos portadores es del 25%; con la misma probabilidad, un hijo de dos portadores no tendrá un defecto genético. En otro 50% de los casos, será portador de AME, pero él mismo no se enfermará.
En casos raros (menos del 2%), los niños afectados nacen en familias donde sólo uno de los padres es portador. En el segundo padre, se produce una mutación genética cuando se pone un óvulo o un espermatozoide.
¿Qué se daña como resultado de una mutación?
Debido a un gen defectuoso en el cuerpo, se altera la producción de la proteína SMN, la proteína de supervivencia de las neuronas motoras. Sin esta proteína, las neuronas motoras, las células nerviosas de la médula espinal responsables de la coordinación de los movimientos y el tono muscular, mueren y la señal no llega a los músculos de las piernas, la espalda y, en parte, los brazos.
Sin el tono necesario, los músculos se atrofian gradualmente. La ausencia de músculos abdominales y de la espalda provoca, entre otras cosas, grandes curvaturas de la columna, lo que provoca problemas respiratorios, que ya existen debido a la debilidad de los músculos.
La enfermedad puede manifestarse desde los primeros meses de vida o en una edad posterior.

¿Qué determina la gravedad de la enfermedad?
Dos genes son responsables de la producción de la proteína SMN: SMN1 y SMN2.
Al mismo tiempo, SMN1 es el principal “cliente” de esta proteína, y SMN2 es uno adicional, produce proteína en una cantidad insuficiente para el funcionamiento normal del organismo. En los casos en que SNM1 está ausente en el genoma humano, SNM2 comienza a realizar funciones de reemplazo, pero nunca puede compensar completamente la deficiencia.
Hay hasta ocho copias de SMN2 en el genoma. La gravedad de la afección del paciente depende de la cantidad de copias de SMN2 que tenga. Un mecanismo tan complejo de la enfermedad lleva al hecho de que la AME tiene varias formas y la condición de los pacientes es muy diferente.
¿Qué formas de AME existen?
Hay 4 tipos de AME, que se diferencian en la gravedad y la edad en la que aparece la enfermedad por primera vez.
AME I, enfermedad de Werdnig-Hoffman. La forma más grave de la enfermedad se manifiesta en bebés de 0 a 6 meses. Los niños con esta forma desde el nacimiento tienen dificultades para respirar, chupar y tragar, y tampoco dominan los movimientos controlados más simples: no mantienen la cabeza erguida ni se sientan solos. Anteriormente se creía que la mayoría (80%) no vivía más allá de los dos años. Ahora, gracias a nuevas estrategias de ventilación mecánica y alimentación por sonda, la vida se puede prolongar varios meses más.
AME II, enfermedad de Dubowitz. Las primeras manifestaciones de la enfermedad se producen entre los 7 y 18 meses. Una persona con este tipo de AME puede comer y sentarse, pero no puede caminar de forma independiente. La esperanza de vida depende del grado de daño a los músculos que proporcionan la respiración.
AME III, Enfermedad de Kugelberg-Welander. La enfermedad aparece por primera vez después de un año y medio. Estos pacientes pueden ponerse de pie (con dolor), pero no caminar. La AME tipo III, por regla general, no afecta la esperanza de vida, pero empeora enormemente su calidad.
AMEIV, este tipo también se denomina “AME del adulto”, ya que la enfermedad suele aparecer después de los 35 años.
Los síntomas incluyen debilidad muscular, escoliosis y temblores. Además, se desarrollan contracturas articulares (movilidad articular limitada) y trastornos metabólicos.
La progresión de la enfermedad no es muy rápida, primero la debilidad muscular afecta los músculos de las piernas y luego los brazos. Por lo general, los pacientes no tienen problemas para tragar ni para la función respiratoria.
La mayoría de los pacientes con AME tipo IV pueden caminar y sólo unos pocos tienen que recurrir a sillas de ruedas.
AME asociada con alteración del gen SMN, en literatura medica Se llaman proximales: constituyen el 95% de todas las amiotrofias espinales. Hay bastantes AME no asociados con el gen SMN, pero son raros. Estos incluyen, por ejemplo, la enfermedad de Kennedy. Los estudios realizados en la década de 1990 demostraron que la enfermedad de Kennedy no está asociada con una degradación del gen SMN1, sino con otras mutaciones genéticas que conducen a una absorción deficiente de la proteína SMN. La enfermedad se manifiesta en personas mayores de 35 años. SMBA se caracteriza principalmente por debilidad de las extremidades.
Un tipo de AME no asociado con el gen SMN se llama La enfermedad de Kennedy. El hecho de que a veces todavía se haga referencia a esta enfermedad como AME es un anacronismo. A finales de la década de 1960, cuando se describió en detalle esta atrofia, se la consideró un tipo de AME porque afecta a los mismos nervios y músculos que los tres tipos de AME (pero en mucha menor medida).
Como es tratado?
Actualmente, no existe un tratamiento radical para la AME.
La corporación internacional Biogen desarrolló el medicamento Spinraza, que mejoró significativamente la condición de los pacientes a quienes se aplicó durante las pruebas. Actualmente, el medicamento está aprobado para su uso en EE.UU.; en Europa, el coste estimado de un curso anual, según estimaciones de la empresa, será de unos 270 mil euros; en Rusia, el medicamento no está certificado. Tratamiento de por vida.
¿Es posible ayudar a los pacientes con AME y cómo exactamente?
Todavía no es posible curar la enfermedad, pero sí es posible aliviar la condición de los pacientes con AME, es decir diferentes caminos compensar los síntomas de la enfermedad.
En los tipos graves de AME, se debe ayudar a los pacientes a respirar y tragar. Por lo tanto, necesitan de manera vital ventiladores móviles, aspiradores de tos y bolsas Ambu.
Los niños con AME también necesitan realmente la ayuda de voluntarios que puedan al menos un tiempo corto reemplazar a los padres.
Los niños con AME pueden necesitar ayuda en cualquier momento, por lo que las mamás y los papás están siempre alerta y dominan las habilidades de reanimación necesarias en caso de que el niño deje de respirar repentinamente.
Los pacientes menos graves necesitan medicamentos para facilitar la respiración, corsés, cochecitos y otros dispositivos que faciliten el movimiento y la vida de las personas con músculos débiles.
Una enfermedad que dura muchos años es agotadora, por lo que los pacientes, especialmente los adultos, suelen necesitar la ayuda de un psicólogo.

El fondo opera en toda Rusia. El trabajo de la fundación tiene dos direcciones principales: brindar asistencia a los pacientes con AME y a sus seres queridos y trabajar para lograr cambios sistémicos en la situación de la AME en Rusia.
Puede apoyar las actividades de la fundación haciendo una donación de la forma que más le convenga. Puedes ayudar haciendo una donación única o periódica en una página especial del fondo o enviando un SMS al número corto 3443 con la palabra SMA y, separado por un espacio, el monto de la donación - por ejemplo, SMA 300.
¿Es posible contraer AME debido a las vacunas?
En Europa y Estados Unidos no se ha rastreado la conexión entre la vacunación y la manifestación de la enfermedad.
Comprender si existe una conexión entre la AME y las vacunas puede ayudar a explicar la diferencia entre la AME y la polio. La poliomielitis es una enfermedad infecciosa cuando la infección daña el cuerpo de un niño inicialmente sano. Un niño con AME, nacido con un genoma dañado, puede parecer saludable en apariencia, pero en realidad ya está enfermo, los síntomas de su enfermedad simplemente aparecen gradualmente. En este sentido, la AME es la misma enfermedad "retardada" que, por ejemplo, la distrofia muscular de Duchenne o el síndrome de Rett, cuando un niño, que se ha desarrollado normalmente durante algún tiempo, pierde habilidades previamente adquiridas y queda discapacitado.
La mayoría de las manifestaciones de AME están asociadas con el desarrollo de las primeras habilidades motoras. Las primeras manifestaciones de la enfermedad coinciden en el tiempo con varias vacunaciones relacionadas con la edad. Como resultado, una persona y su familia pueden afirmar que "se enfermó por la vacuna", pero en realidad simplemente mostró signos de una enfermedad que ya existía.
¿Cómo se determina que un niño tiene AME y no alguna otra enfermedad?
A pesar de que la AME fue descrita por primera vez por el neurólogo austriaco Guido Werdnig y el neurólogo alemán Johann Hoffmann a principios de la década de 1890, la naturaleza de la enfermedad no se comprendió plenamente hasta finales del siglo XX. El gen SMN1 fue descubierto en 1995. Para confirmar el diagnóstico de AME, se necesita una prueba genética.
En Rusia, las pruebas genéticas apropiadas estuvieron disponibles a principios de la década de 2000. Se puede realizar una prueba genética de AME bajo el seguro médico obligatorio, pero en la práctica, no muchos médicos conocen este diagnóstico poco común y derivan a los pacientes para el estudio adecuado. El coste de estas pruebas en laboratorios comerciales de Moscú es de unos 6 mil rublos.
La falta de diagnósticos específicos también ha generado confusión en los diagnósticos. La mayoría de los pacientes con AME en Rusia no son identificados; muchos de los identificados tienen registrado como diagnóstico la “enfermedad de Werdnig-Hoffmann”, aunque no todos (especialmente los adultos) realmente padecen este tipo de enfermedad.
¿Cuántos pacientes con AME hay en Rusia?

El medicamento "Spinraza", que mejora significativamente la condición de los pacientes. Foto de healthbeat.spectrumhealth.org
Teniendo en cuenta la frecuencia de la enfermedad, el número de pacientes con AME en Rusia debería ser de siete a veinticuatro mil personas. Hoy en día, hay alrededor de 400 personas en el registro de pacientes de la SMA Families Foundation.
¿Quién en Rusia ayuda a las personas con AME y a sus familias?
Fundación benéfica "Vera", hospicio infantil "Casa con faro", fundación benéfica "Paliativo infantil", fundación benéfica "Familias de SMA", servicio paliativo infantil "Mercy".
Desde 2014, se desarrolla en Moscú un proyecto conjunto del servicio "Mercy" y la fundación "Familias de AME" "SMA Clinics". En reuniones que tienen lugar una vez al mes, los pacientes pueden recibir consultas de un neumólogo, ortopedista y fisioterapeuta. y psicólogo. Recientemente, algunas reuniones se han centrado en las necesidades de los pacientes adultos.
Personajes famosos con AME

La italiana Simona Spinoglio. Nació con una enfermedad hereditaria: atrofia muscular espinal tipo 2. Desde que nació no puede caminar y se mueve únicamente con la ayuda de una silla de ruedas eléctrica. Pero su vida es plena y llena de acontecimientos; nada puede detener su deseo de vivir.
Simone trabaja en la línea directa de la Asociación Italiana de Familias de AME y ayuda a niños y adultos con AME y otras enfermedades neuromusculares.
Simone también grabó varias canciones populares en la comunidad italiana de AME, sobre la libertad de hacer lo que quieras, a pesar de la enfermedad.
La cantante rusa Yulia Samoilova nacida en la ciudad de Ukhta (República de Komi). A la edad de diez años actuó en un concierto benéfico, tras lo cual fue invitada a estudiar canto en el Palacio de los Pioneros local. A los quince años comenzó a estudiar en la Casa de Cultura de la ciudad.
En 2008 formó su propio grupo musical (se separó en 2010). En 2013, participó en el concurso "Factor A" del canal de televisión Rossiya. Obtuvo el segundo lugar y recibió el premio personal de Alla Pugacheva "Alla's Golden Star". En 2017, debido a la exclusión de Rusia de programa competitivo No pudo participar en el Festival de la Canción de Eurovisión. Se desplaza en silla de ruedas.
Programador de Vladimir Valery Spiridonov. Se graduó de la escuela con una medalla de oro y luego defendió su título de ingeniero. En 2015, Valery planeaba participar en un experimento del cirujano italiano Sergio Canavero para trasplantar una cabeza humana (el experimento fue cancelado).
Hoy Valery es miembro de la Cámara Pública de la ciudad de Vladimir, experto en cuestiones de entorno accesible y creador de su propia comunidad "Deseo de vida", que habla sobre la creación de un entorno accesible y proyectos médicos prometedores. Valery participa en muchos programas de televisión de la televisión rusa y extranjera.
Atrofia muscular en la columna(AME), o amiotrofia, Es una enfermedad hereditaria que se acompaña de trastornos agudos en la actividad de las neuronas de la médula espinal y el cerebro. Los procesos afectan a las neuronas motoras. La enfermedad fue descrita por primera vez según el cuadro médico en el siglo XIX. Pertenece a un grupo de trastornos genéticos causados por mutaciones.
La especificidad de la atrofia muscular radica en el hecho de que solo un tipo de patología de la columna, la primera, se desarrolla en un recién nacido entre 1 y 2 meses de vida. Otras formas de la enfermedad se hacen sentir sólo en la edad adulta. La forma compleja de atrofia espinal y los métodos de su tratamiento se estudian en disciplinas como la genética, la neurología y la pediatría.
Hay diferentes estimaciones de qué tan común es. atrofia muscular en la columna en niños recién nacidos. La densidad de casos está directamente relacionada con la población de un lugar particular del planeta. Debido a que la patología a menudo se descubre sólo en la edad adulta, el número de casos después de 20 años es mayor que en la infancia. Aproximadamente 1 de cada 20.000 personas padece alguna forma de este trastorno.
¡Hecho! Entre los bebés, las formas graves de enfermedades de la columna ocurren en promedio entre 5 y 7 veces por cada 100.000 personas.
El factor hereditario no se manifiesta en todos. Por tanto, los padres pueden ser portadores de un gen mutado. Pero sólo aparecerá en un niño con una probabilidad del 50-70%. Se cree que la prevalencia de AME entre los portadores es de 1 en 80 familias, o en 160 personas de diferentes sexos.
La AME es una de las formas más comunes de procesos degenerativos hereditarios en los niños. Ocupa el segundo lugar después de la fibrosis quística y se considera la causa número uno entre las enfermedades hereditarias que provocan la muerte de un niño antes de que cumpla entre 15 y 18 años.

La muerte se produce por insuficiencia respiratoria. Cuanto antes se manifieste la patología de la columna, peor será el pronóstico. En promedio, los niños con atrofia musculoesquelética espinal viven entre 10 y 11 años. Al mismo tiempo, el estado de inteligencia no afecta de ninguna manera el progreso de la amiotrofia espinal.
El trastorno se presenta con más frecuencia en niños que en niñas, y en ellos es mucho más complejo. Por cada 1 paciente mujer hay 2 pacientes hombres. Pero a partir de los 8 años la frecuencia entre las niñas aumenta.
Factores genéticos de la enfermedad.
La atrofia muscular espinal ocurre cuando se hereda el genoma recesivo del cromosoma 5. Si ambas personas que dieron a luz a un bebé son portadoras de AME, entonces existe al menos un 25% de posibilidades de transmitir el gen al niño. Como resultado, se altera la síntesis de estructuras proteicas y la destrucción de las neuronas motoras de la médula espinal ocurre varias veces más rápido que la restauración.
Durante el desarrollo embrionario, el sistema nervioso del niño produce sólo la mitad del volumen necesario de neuronas motoras. Con el tiempo, con la AME, este proceso se ralentiza mucho. Después del nacimiento, se desarrolla atrofia espinal debido a la falta de estructuras.

Características del funcionamiento de las neuronas.
Un cerebro activo envía constantemente impulsos a médula espinal, y las células nerviosas sirven como conductoras. Envían señales a los músculos, lo que da como resultado su movimiento. Si este proceso se interrumpe, el movimiento se vuelve imposible.
Para la atrofia muscular espinal neuronas motoras Las piernas, que forman parte de la médula espinal, no funcionan correctamente. Son responsables de las señales que ayudan al cerebro a respaldar funciones como gatear, sostener el cuello, apretar y mover brazos y piernas, así como la respiración y el reflejo de deglución.
¡Importante! Al recibir copias defectuosas del gen SMN1 de los padres, el sistema nervioso del niño deja de producir una proteína que controla el proceso de síntesis y metabolismo de las neuronas.

Como resultado, los músculos que no reciben señales constantes comienzan a atrofiarse.
Clasificación de tipos de atrofia.
Hay 4 grupos comunes de atrofia muscular espinal en niños y adultos:
- Forma infantil. Mayoría tipo complejo Atrofia musculoesquelética espinal, también llamada patología de Werdnig-Hoffmann. El curso de la patología en esta forma se complica por el rápido desarrollo de síntomas graves: dificultades para tragar, chupar y respirar. Los bebés con AME1 no pueden mantener la cabeza erguida ni sentarse normalmente.
- Forma intermedia. La AME2, o enfermedad de Dubowitz, difiere ligeramente en gravedad. Con esta forma de patología, el niño puede permanecer sentado e incluso comer, ya que las funciones de deglución están parcialmente intactas. Pero no podrá caminar. El pronóstico está directamente relacionado con el grado de daño a los músculos respiratorios responsables de la actividad de los pulmones.
- Uniforme juvenil. La AME3, o enfermedad de Kugelberg-Welander, es más fácilmente tolerada por los adolescentes que los primeros tipos de atrofia muscular espinal. El niño puede ponerse de pie, pero sufrirá una debilidad grave. El riesgo de discapacidad es alto: la mayoría todavía necesita una silla de ruedas.
- Tipo adulto. SMA4 ocurre principalmente después de 35 años. La esperanza de vida con la enfermedad no cambia, pero el paciente desarrolla debilidad muscular grave y disminución de los reflejos tendinosos. A medida que avanza, se requiere una silla de ruedas.

Es muy difícil sospechar una patología espinal-muscular inmediatamente después del nacimiento. Pero la detección temprana puede ayudar a aliviar el sufrimiento de los pacientes, por lo que es necesario estar atento a los síntomas comunes de la atrofia muscular espinal.
Síntomas de diferentes formas de la enfermedad.
Existe un conjunto general de signos de AME que pueden usarse para sospechar patología si no se detectan otros problemas o el diagnóstico es dudoso. El grupo de síntomas se reduce a la manifestación de parálisis periférica fláccida:
- debilidad muscular severa o atrofia de diferentes grupos de músculos;
- primero, las extremidades están involucradas en el proceso: simétricamente, las piernas y luego los brazos, gradualmente también se involucra el torso;
- no hay trastornos de sensibilidad ni trastornos pélvicos;
- los problemas más pronunciados afectan a los grupos de músculos proximales o distales.

Los pacientes experimentan espasmos y fibrilación: fibrilación auricular.
Signos de AME1
Hay 3 tipos de enfermedad de Werdnig-Hoffman:
- Forma congénita. Comienza entre 1 y 6 meses de vida y tiene síntomas graves. Se pueden detectar signos durante el desarrollo intrauterino: el embrión se moverá poco. La hipotensión se observa inmediatamente después del nacimiento del niño. Estos bebés no pueden mantener la cabeza erguida ni sentarse. Están constantemente en postura de rana con las extremidades abiertas. Los síntomas aparecen primero en las piernas, luego en los brazos, tras lo cual los músculos respiratorios sufren. El desarrollo mental de estos niños es lento y rara vez viven más de 2 años.
- Atrofia muscular espinal temprana. Los primeros signos comienzan a molestar al paciente antes de 1,5 años, con mayor frecuencia después de cualquier infección. Aunque antes el niño podía estar de pie y sentado, ahora pierde estas funciones. Se desarrolla paresia y luego los músculos respiratorios se ven afectados. El niño muere, por regla general, como resultado de una neumonía prolongada o insuficiencia respiratoria a la edad de 3 a 5 años.
- Forma tardía. La patología ocurre después de 1,5 años, las habilidades motoras se conservan en un niño hasta los 10 años. La lenta progresión de los síntomas conduce a insuficiencia respiratoria y muerte antes de los 18 años.

La AME1 es la forma más grave de la patología; siempre hay que prepararse para el peor resultado.
Signos de la enfermedad de Kugelberg-Welander
Ocurre entre las edades de 2 y 15 años. Primero, las extremidades inferiores están involucradas en el proceso, luego la cintura pélvica, en las etapas finales, la cintura escapular y el sistema respiratorio sufren. Aproximadamente el 25% de los pacientes desarrolla el síndrome de pseudohipertrofia muscular, por lo que la patología se confunde con la enfermedad muscular de Becker.
La atrofia muscular espinal de Kugelberg-Welander no se acompaña de deformidades óseas y los pacientes pueden cuidar de sí mismos durante muchos años.

Amiotrofia Kennedy
Esta patología se incluye en el grupo de adultos, afectando a varones a partir de los 30 años. Las mujeres no padecen la patología. El curso es moderado, los músculos de las piernas se ven afectados primero y durante los siguientes 10 a 20 años el paciente mantiene el ritmo de vida habitual. Sólo después de esto los músculos de los brazos y la cabeza comienzan a sufrir. Muchos pacientes experimentan cambios endocrinos con el tiempo: atrofia testicular, falta de libido, diabetes mellitus.

ACM distal
Esta forma de atrofia muscular espinal también se desarrolla en pacientes adultos después de los 20 años. Su segundo nombre es Duchenne-Arana SMA. El riesgo de desarrollar patología persiste hasta los 50 años. La atrofia comienza en las manos, causando el síndrome del pie en garra, y luego pasa a los músculos grandes. Con el tiempo, aparece paresia de los músculos de las extremidades inferiores y el torso rara vez sufre. El pronóstico para esta forma es favorable a menos que se asocie distonía de torsión o enfermedad de Parkinson.

AME Vulpiana
Forma escapuloperonea de atrofia muscular espinal, acompañada del síntoma de omóplatos "alados". Aparece en promedio entre los 20 y los 40 años, luego ocurre con menos frecuencia. La cintura escapular se ve afectada y, después de un tiempo, los brazos y las extremidades inferiores. Con esta forma de enfermedad de la columna, las funciones motoras del paciente se conservan durante 30 a 40 años.

Métodos para diagnosticar patología.
Es posible reconocer la atrofia muscular espinal con una garantía del 100% sólo con la ayuda de Análisis de ADN para factores genéticos moleculares. Con su ayuda, puedes encontrar un gen defectuoso en el cromosoma 5.
También usado análisis bioquímico para identificar el estado de la proteína. Es necesario un estudio electrofisiológico del cerebro para determinar la actividad de los impulsos y los troncos nerviosos. Rara vez se prescriben MRI y CT, ya que estos métodos no son muy efectivos.

Métodos de tratamiento
No existen tratamientos eficaces para la atrofia muscular espinal. Sin embargo, las etapas leves se pueden corregir. La fisioterapia, los masajes y los medicamentos pueden ayudar a que su hijo se sienta cómodo. En la edad adulta, la terapia es más eficaz, ya que estas formas de atrofia no son tan difíciles de tolerar.
Medicamentos
Para corregir el trabajo de las fibras musculares y los impulsos nerviosos se utilizan fármacos que mejoran la circulación sanguínea y ralentizan la destrucción de las neuronas:
- Anticolinesterasa. Los medicamentos suprimen la actividad de la enzima que descompone la acetilcolina: "Proserin", "Oxazil", "Sangviritrin".
- Vitaminas y suplementos dietéticos. Utilizan antioxidantes, carnitina y vitaminas B para mantener el metabolismo y el tono.
- Nootrópicos. Mejorar el funcionamiento del sistema nervioso: Nootropil, Kaviton, Semax.
- Medios para activar el metabolismo. Este grupo incluye varios productos: ácido nicotínico, Actovegin, orotato de potasio.

También es importante apoyar una nutrición adecuada del niño y evitar el abuso de grasas y alimentos refinados.
Fisioterapia
Los procedimientos fisioterapéuticos para la atrofia muscular espinal mejoran el tono, la circulación sanguínea, el metabolismo y ayudan a reducir el dolor. Prescrito: UHF, electroforesis, técnicas manuales, aparatos respiratorios para estimular los pulmones.
Control cuidadoso de la respiración
Dado que la atrofia muscular espinal a menudo se asocia con trastornos como la respiración, es necesario controlar estrictamente el funcionamiento de este sistema en el niño:
- se prescribe fisioterapia pecho;
- limpia el tracto respiratorio de la mucosidad que se forma;
- se recetan analgésicos;
- tomar medicamentos que reduzcan la producción de secreciones;
- utilizar métodos de ventilación no invasivos que aumenten la comodidad del paciente y prevengan la hipoventilación nocturna;
- Se utilizan métodos invasivos: ventilación artificial mediante un tubo insertado.

Este último método se utiliza en casos graves cuando el reflejo respiratorio se vuelve imposible.
Nutrición infantil
Si la atrofia muscular espinal se ha desarrollado hasta el punto de que el paciente ya no puede tragar por sí solo, necesita ayuda externa. Es necesario corregir la debilidad muscular.
Un médico especialista en atrofia muscular habla en detalle sobre cómo alimentar a un pequeño paciente con disfunción para tragar. A veces se requiere asistencia médica profesional para lograr estos objetivos.
¡Importante! Al tratar a pacientes con AME, no es necesario seguir una dieta estricta ni introducir/restringir alimentos que contengan determinadas sustancias, vitaminas y minerales.
En los niños con AME, el proceso de digestión puede verse alterado, por lo que los niños comienzan a sufrir estreñimiento. A veces se desarrolla la enfermedad por reflujo.
Previsión y posibles consecuencias.
Si se detecta atrofia muscular espinal en un paciente en la edad adulta, el pronóstico es más favorable. La patología SMA1 rara vez deja esperanza – La mayoría de los niños no viven hasta los 2 años, el resto muere antes de los 5 años.
La muerte se produce por insuficiencia respiratoria y, con menos frecuencia, por neumonía aguda y persistente. Actualmente, no existen formas de prevenir la enfermedad.
Los adultos diagnosticados con AME deben abandonar los malos hábitos, los deportes extremos y los horarios irregulares de descanso y trabajo. Esto ralentizará significativamente la progresión de la enfermedad muscular de la columna.
Atrofia muscular en la columna(AME): enfermedades degenerativas con daño a las neuronas motoras que se inician en el período prenatal y tienen un curso progresivo en la infancia y infancia. La denervación muscular progresiva se compensa en parte con la reinervación de unidades motoras adyacentes, creando así unidades motoras gigantes, y se desarrolla una mayor atrofia de las fibras musculares cuando las neuronas reinervadas finalmente participan en el proceso patológico. Las neuronas motoras centrales no se ven afectadas.
Atrofia muscular en la columna(AME) se dividen en forma infantil grave (enfermedad de Werdnig-Hoffmann o AME tipo 1), forma infantil tardía, forma progresiva más lenta (AME tipo 2) y forma juvenil con más curso largo(Enfermedad de Kugelberg-Welander o AME tipo 3). Esta clasificación se basa en la presentación clínica, incluida la edad de inicio del paciente, la gravedad de la debilidad muscular y el curso de la enfermedad. La biopsia muscular no diferencia entre los tipos 1 y 2, aunque el tipo 3 tiene un patrón de denervación-reinervación adulto más que infantil.
Algunos pacientes según la clínica. manifestaciones ocupan una posición intermedia entre los tipos 1 y 2 o entre los tipos 2 y 3. Una variante de la atrofia muscular espinal (AME), la enfermedad de Fazio-Londe, es una enfermedad progresiva. parálisis bulbar, resultante de la degeneración de las neuronas motoras que es más grave en el tronco del encéfalo que en la médula espinal.
Razón espinal(SMA) sirve como la ausencia de detención en una determinada etapa del desarrollo de la muerte celular programada, que es un fenómeno normal en el período embrionario. El neuroectodermo primitivo produce una gran cantidad de neuroblastos motores y otras neuronas, pero sólo alrededor del 50% de ellos sobrevive y madura hasta convertirse en neuronas. Ciclo vital Las células “sobrantes” son limitadas y sufren cambios degenerativos.
Si en cierto etapas el proceso que detiene la muerte fisiológica de las células no se desarrolla, la muerte de las neuronas puede continuar en el período fetal tardío y posnatal. El gen de supervivencia de la neurona motora (SMN) detiene la apoptosis de los neuroblastos motores. A diferencia de la mayoría de los genes, que se conservan a través de la evolución, SMN es un gen exclusivo de los mamíferos.
Manifestaciones clínicas de la atrofia muscular espinal (AME)
Principal signos de atrofia muscular espinal(AME) tipo 1 son hipotensión severa, debilidad muscular generalizada, disminución de la masa muscular, ausencia de reflejos tendinosos, afectación de los músculos de la lengua, la cara y los músculos masticatorios, preservación de la función de los músculos extrínsecos del ojo y los esfínteres. Los bebés con manifestaciones clínicas de la enfermedad al nacer pueden experimentar problemas respiratorios e incapacidad para comer.
Las contracturas congénitas, cuya gravedad varía desde un simple pie zambo hasta una artrogriposis generalizada, ocurren en aproximadamente el 10% de los recién nacidos gravemente afectados. Los bebés se encuentran en una posición relajada de rana, la actividad motora espontánea se reduce, los niños no pueden superar la fuerza de gravedad de sus extremidades y no pueden mantener bien la cabeza erguida. Más de 2/3 de los niños que padecen esta enfermedad mueren antes de los 2 años, en muchos casos la muerte se produce en la primera infancia.
Bebés con atrofia muscular espinal(AME) tipo 2 generalmente pueden succionar y tragar, y la función respiratoria no se ve afectada en la primera infancia. A pesar de la debilidad muscular progresiva, muchos pacientes sobreviven hasta la edad escolar y más allá, aunque en las últimas etapas de la enfermedad hay una discapacidad grave y los pacientes están confinados a una silla de ruedas. El tono de voz nasal y la dificultad para tragar aparecen a mayor edad. En muchos pacientes con una larga esperanza de vida, la escoliosis se convierte en una de las principales complicaciones de la enfermedad.
Enfermedad de Kugelberg-Welander- mayoría forma ligera atrofia muscular espinal (AME) (tipo 3). En la infancia manifestaciones clínicas las enfermedades pueden estar ausentes. Se desarrolla debilidad progresiva en las extremidades proximales, especialmente en los músculos de la cintura escapular. Los pacientes conservan la capacidad de caminar de forma independiente. Rara vez aparecen síntomas de debilidad de los músculos bulbares.
En aproximadamente el 25% de los pacientes con esta forma atrofia muscular en la columna(SMA) la hipertrofia muscular es más pronunciada que la atrofia muscular; por lo tanto, la distrofia muscular puede diagnosticarse erróneamente. Los pacientes pueden sobrevivir hasta la edad adulta. Las fasciculaciones son un signo clínico específico de denervación muscular. En niños delgados, se pueden encontrar fasciculaciones en el deltoides, el bíceps braquial y, en ocasiones, el cuádriceps femoral. Sin embargo, los constantes movimientos involuntarios parecidos a los de un gusano pueden quedar enmascarados por una gruesa capa de grasa subcutánea. Las fasciculaciones se identifican mejor en la lengua, donde prácticamente no existe tejido conectivo subcutáneo que separe la capa muscular del epitelio. Cuando el músculo de la lengua se contrae, por ejemplo al llorar o al sacar la lengua, las fasciculaciones son menos visibles que en una lengua relajada.
Al estirar los brazos hacia adelante en niños con atrofia muscular en la columna(AME) suele haber un temblor característico en los dedos debido a la fasciculación y la debilidad muscular. Este temblor no debe confundirse con el temblor debido a lesiones cerebelosas. La mialgia no es típica de la AME.
Para la atrofia muscular espinal(AME) no desarrolla daño cardíaco. La inteligencia se conserva y, a menudo, los niños con atrofia muscular espinal (AME) muestran capacidades intelectuales más altas que sus compañeros sanos, ya que la energía que no puede dirigirse a la actividad física se realiza en la esfera de desarrollo intelectual. Además, estos niños tienen más probabilidades de interactuar con adultos que con niños debido a las restricciones sociales causadas por la enfermedad.
La AME es una enfermedad hereditaria causada por una mutación genética. Conduce a la muerte de las neuronas motoras que controlan el movimiento de los músculos esqueléticos. Esta muerte es la causa de la atrofia de los músculos de las piernas, la cabeza y el cuello, lo que conduce a una alteración de los movimientos voluntarios (gatear, tragar, sostenerse la cabeza, caminar). Es posible que los músculos del brazo no se vean afectados.
Dependiendo de la gravedad y el estado de salud, existen 3 tipos de atrofia muscular espinal
AME tipo I (Werdnig-Hoffmann):
Este tipo de enfermedades se pueden detectar durante el embarazo o durante los primeros meses posteriores al mismo.
Un niño que padece esta afección se moverá poco y débilmente durante el embarazo, y al nacer experimentará síntomas como debilidad y atrofia de los músculos de brazos y piernas, además de dificultad para tragar, un reflejo de succión débil y dificultades respiratorias importantes.
Este tipo de AME es, con diferencia, el más grave. La mayoría de los niños que padecen este tipo de atrofia muscular espinal corren el riesgo de morir durante los primeros años después del nacimiento.
AME tipo II
El tipo intermedio de la enfermedad se manifiesta en el primer año y medio de vida.
En la enfermedad de tipo II, los músculos de las piernas sufren más y la enfermedad progresa más lentamente.
Los pacientes que padecen este tipo de AME no pueden pararse ni sentarse sin ayuda. Causa: debilidad de los músculos de las piernas y del suelo pélvico.
La muerte es posible después de los 2 años, pero el pronóstico es más favorable que en la enfermedad de tipo I.
AME tipo III (Kugelberg-Welander)
La atrofia muscular espinal tipo III aparece entre los 2 y los 5 años de edad, pero tiene riesgo de aparecer entre los 2 y los 17 años.
Este tipo de AME progresa muy lentamente. Al inicio de la enfermedad, el paciente puede ponerse de pie y caminar sin ayuda, pero con el tiempo tiene dificultades para caminar, mantener la posición sentada y ponerse de pie. Poco a poco la enfermedad se extiende a las extremidades superiores.
El pronóstico para la AME tipo III es el más favorable. Pero debido al número relativamente pequeño de pacientes que padecen esta enfermedad (aproximadamente 4 personas de cada persona), se han realizado pocas investigaciones. Lo bueno es que recientemente su número ha aumentado significativamente, lo que significa que ha aumentado la probabilidad de encontrar una cura para esta enfermedad tan grave.
Hoy no hay tratamiento y terapia efectiva, ayudando a restaurar las neuronas dañadas. Pero para detener temporalmente los síntomas de la atrofia muscular espinal, un paciente que padece esta afección necesita apoyo para la función de drenaje del tracto respiratorio, fisioterapia, corrección ortopédica y vitaminas B, vitamina E. Esto ayudará a fortalecer los músculos y mantener su función durante el mayor tiempo posible.
Síntomas y tratamiento de la atrofia muscular espinal.
La atrofia muscular espinal (AME), o amiotrofia espinal de Werdnig-Hoffmann, es una enfermedad hereditaria autosómica recesiva que se caracteriza por hipotensión progresiva y debilidad muscular.
El característico debilitamiento del tejido muscular se produce debido a la degeneración progresiva de las neuronas motoras alfa en el asta anterior de la médula espinal. Así, la enfermedad se basa en una patología de la médula espinal, que puede heredarse.
Una característica de la enfermedad es una manifestación más activa de debilidad en los músculos esqueléticos ubicados más profundamente que los ubicados más cerca de la superficie del cuerpo. En este material hablaremos sobre los síntomas y el tratamiento de la atrofia muscular espinal de Werdnig-Hoffmann.
La información será útil para cualquiera que, por diversas razones, haya tenido que lidiar con esta enfermedad grave y a menudo mortal.
Características y tipos del síndrome AME.
En algunos pacientes, las neuronas motoras de los pares craneales, especialmente de los pares V a XII, también pueden estar involucradas en el proceso patológico. En este caso, la enfermedad se origina en cuerno posterior células de la médula espinal, que además provoca insuficiencia de los músculos del diafragma, tracto gastrointestinal, corazón y esfínteres.
En 1890, Werdnig describió por primera vez la forma infantil clásica de AME, una manifestación del síndrome en niños pequeños. Muchos años después, en 1956, Kugelberg y Welander clasificaron una forma menos grave de atrofia muscular espinal en pacientes de edad avanzada.
Gracias a estos científicos, hoy los médicos pueden diferenciar con precisión la AME de diferentes tipos de enfermedades con síntomas similares, por ejemplo, la distrofia muscular de Duchenne.
La amiotrofia espinal es el diagnóstico más común en las niñas, con debilidad severamente progresiva. Es una de las causas genéticas más comunes de muerte en niños.
El síndrome de AME se divide en cuatro tipos según la edad del paciente de la siguiente manera:
- Tipo I (amiotrofia espinal de Werdnig-Hoffmann). Se desarrolla hasta los 6 meses de edad.
- Tipo II – entre 6 y 12 meses de edad.
- Tipo III (enfermedad de Kugelberg-Welander): de 2 a 15 años
- Tipo IV en pacientes adultos.
Extensión
La incidencia de la enfermedad es de aproximadamente un caso por cada mil personas. Si hablamos solo de recién nacidos, esta cifra será de aproximadamente 5 a 7 casos por cada 100 mil. Dado que la amiotrofia espinal es una enfermedad hereditaria recesiva, muchos padres pueden ser portadores de ella y no saberlo.
La prevalencia de portadores de AME es de 1 entre 80, es decir, una de cada 80 familias puede tener un hijo que padezca atrofia muscular espinal. Este riesgo aumenta varias veces cuando ambos padres son portadores del gen mutante.
Así, el síndrome de AME es la enfermedad degenerativa del sistema nervioso más común en niños después de la fibrosis quística y, como se señaló anteriormente, es la principal causa hereditaria mortalidad infantil.
La muerte se debe a insuficiencia respiratoria. Cuanto más joven es el paciente primeras etapas enfermedad, peor es el pronóstico. La edad promedio general al momento de la muerte es de unos 10 años. El estado de inteligencia y otros indicadores del desarrollo psicosomático del niño no afectan en modo alguno la progresión de la enfermedad.
A diferencia de los pacientes jóvenes, los hombres adultos se ven afectados con mayor frecuencia por la enfermedad que las mujeres en una proporción de aproximadamente 2:1, mientras que el curso clínico en los pacientes masculinos es más grave. El aumento de casos de la enfermedad en las mujeres comienza alrededor de los 8 años de edad y los niños “alcanzan” a las niñas a los 13 años.
Atrofia muscular espinal - síntomas
El primer tipo de atrofia muscular espinal hace que los primeros síntomas aparezcan antes de que nazca el bebé. La mayoría de las madres informan inactividad fetal anormal durante las últimas etapas del embarazo. Los síntomas de la AME en los recién nacidos son bastante obvios: el niño no puede darse la vuelta por sí solo y posteriormente sentarse.
Además, se desarrolla un deterioro clínico progresivo que en la gran mayoría de los casos termina en la muerte. La muerte suele producirse por insuficiencia respiratoria y sus complicaciones en pacientes a partir de los 2 años.
Los pacientes con AME tipo 2 se desarrollan normalmente durante los primeros 4 a 6 meses de vida. Es posible que puedan sentarse por sí solos, pero nunca podrán caminar y necesitarán una silla de ruedas para desplazarse en el futuro. Por regla general, estos niños viven mucho más tiempo que los pacientes que padecen atrofia espinal de Werdnig-Hoffmann. La esperanza de vida promedio es de hasta 40 años.
Los pacientes con diabetes tipo 3 suelen tener dificultades para subir escaleras o levantarse del suelo, principalmente debido a la debilidad de los extensores. articulación de cadera. La esperanza de vida es cercana a lo normal.
Lea más sobre los síntomas de la AME
Los recién nacidos con atrofia muscular espinal tipo 1 están inactivos. Mueven sus extremidades con gran dificultad, si es que pueden hacerlo. Las caderas están casi constantemente dobladas, debilitadas y se pueden torcer fácilmente con la mano. lados diferentes. Las rodillas también están dobladas.
Debido a que los músculos externos suelen verse menos afectados, los dedos de manos y pies se mueven casi con normalidad. Los bebés no pueden controlar ni levantar la cabeza. La arreflexia (falta de reflejos) se observa en casi todos los pacientes.
Los niños que padecen el segundo tipo de AME pueden mover la cabeza y el 75% de estos pacientes pueden sentarse de forma independiente. La debilidad muscular es mayor en las extremidades inferiores que en las superiores. El reflejo de la rótula está ausente. Los niños mayores pueden demostrar reflejos bíceps y tríceps.
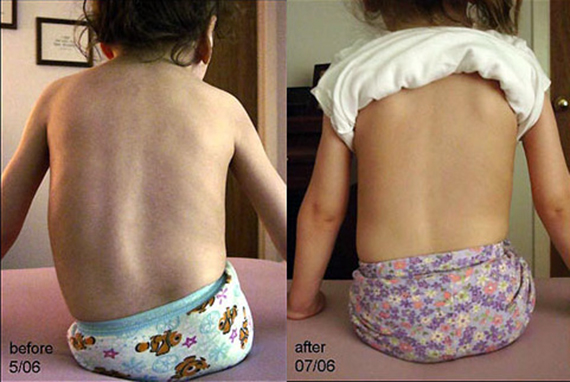
La escoliosis es el síntoma más común de la AME y la mayoría de los pacientes también desarrollan luxación de cadera, ya sea unilateral o bilateral. Estos signos se desarrollan antes de los 10 años.
Los pacientes con enfermedad de atrofia muscular espinal tipo 3 pueden caminar a una edad temprana y esta capacidad se puede mantener de forma ambulatoria durante la adolescencia. La debilidad puede provocar la caída del pie y una resistencia limitada en el cuerpo. Un tercio de los pacientes quedan en silla de ruedas a los 40 años.
Tratamiento
Actualmente no se conoce ningún tratamiento médico para la atrofia muscular espinal, por lo que vale la pena señalar que la tasa de supervivencia entre los pacientes de edad temprana y mediana es bastante baja.
Los recién nacidos con atrofia muscular espinal de Werdnig-Hoffmann requieren pocos cuidados ortopédicos debido a su corta esperanza de vida. La ferulización se utiliza en casos de fracturas, que a menudo se observan con una actividad muscular debilitada.
Para los pacientes con AME tipo II y III, se puede utilizar la fisioterapia para tratar la contractura articular (movimiento articular limitado). Para un tratamiento más radical de la contractura, está indicado el tratamiento quirúrgico.
Como se señaló anteriormente, el problema ortopédico más común en el síndrome de AME es la escoliosis, que a menudo adopta una forma grave. La progresión de la curvatura de la columna es de unos 8° por año, a pesar del tratamiento con aparatos ortopédicos.
La fusión espinal posterior de tipo segmentario a menudo se recomienda para pacientes jóvenes cuya curvatura de la columna vertebral no se puede corregir con aparatos ortopédicos, así como para pacientes mayores de 10 años con una curvatura de más de 40°.
La cirugía debe retrasarse hasta punto medico Esto es posible. Hay que tener en cuenta que la progresión de la curvatura se produce más lentamente en pacientes con el tercer tipo de AME y aparece con mayor frecuencia a una edad más avanzada.
Dieta
El tratamiento de la atrofia muscular espinal requiere un enfoque individual para racionar el menú del paciente, lo que, lamentablemente, muy a menudo no es observado por los médicos tratantes. Se incluyen indicadores antropométricos, composición sanguínea y marcadores bioquímicos de la condición muscular. elementos importantes Evaluaciones en pacientes con AME.
El curso específico de la enfermedad en un paciente en particular puede requerir una intervención en su dieta para influir en los indicadores anteriores, ya que es con la ayuda de los alimentos que los músculos pueden recibir los nutrientes que el paciente necesita en su caso.
Por supuesto, este enfoque del tratamiento amiotrofia muscular espinal es relevante sólo al diagnosticar la segunda o tercera forma de la enfermedad.
Terapia física
El apoyo adicional para un paciente que sufre de atrofia muscular espinal del segundo y tercer tipo no es menos importante que la dieta. En primer lugar, con la ayuda de una carga normalizada, es posible prevenir la progresión de la contractura articular, así como mantener la fuerza, la resistencia y la independencia en el autocuidado.
El ejercicio físico juega un papel bastante importante en los aspectos educativos, sociales, psicológicos y actividad profesional el paciente, ya que tendrá la oportunidad de llevar un estilo de vida casi normal, como las personas sanas.
Síntomas y tratamiento de la artrosis de la columna cervical.
Consecuencias de una fractura de columna
Síntomas y tratamiento de tumores de columna y médula espinal.
Causas del dolor lumbar que se irradia a la pierna.
¿Qué es la espondilitis anquilosante? Síntomas, tratamiento y pronóstico.
Ya hay 1 reseña para este artículo. ¿¿Qué opinas??
Da miedo imaginar cuantos problemas puede tener una persona con su salud, aportas información valiosa, está todo muy claro.
Cancelar respuesta
somnólogo
Ruslán
Puerto pequeño
Zdravo-Bravo, salud, estilo de vida saludable © 2018. Respete los derechos de autor; ¡el uso de los materiales del sitio sólo es posible con un enlace activo a la fuente!
Tipos de AME
La AME relacionada con SMN, o 5qSMA, o AME proximal generalmente se divide en tres categorías. Convencionalmente, también podemos distinguir SMA0 (cero) y SMA4. Por lo tanto, existen varios tipos principales de AME y todos ocurren de manera diferente. El proceso puede desarrollarse en diferentes períodos de la vida y tener sus propios características clínicas, su naturaleza del curso, pronóstico y la cantidad requerida de ayuda y apoyo.
El tipo 1 es el más grave, con el inicio más temprano, el tipo 3 es el menos grave, con una edad de inicio tardía. Algunos expertos también identifican el tipo 4 para designar AME moderada o leve que comienza en la edad adulta.
La especificidad de la enfermedad es que cada niño, incluso dentro del mismo grupo, desarrolla AME de forma diferente e individual. Exteriormente, esto se manifiesta en la posible variedad de movimientos: algunos niños pueden mantener la cabeza erguida, levantar ligeramente los brazos y levantar las piernas, mientras que otros simplemente se acuestan en la clásica posición de "rana" y solo pueden mover ligeramente los pies y dedos.
Victor Dubovits, una de las luminarias en el campo de la gestión de la AME, propuso en los años 90 utilizar una escala más complicada además de la clasificación habitual. Por ejemplo, existe SMA1 y sus subtipos serán 1.1, 1.2, 1.3, es decir. de 1,1 a 1,9. Este esquema se utiliza en Italia.
El sistema americano se basa en la escala ABC, siendo B el tipo clásico, A el tipo débil y C el tipo más fuerte. El sistema con subtipos ABC describe claramente el cuadro clínico de la AME y le permite seleccionar la terapia de mantenimiento para un niño según el subgrupo y el pronóstico correspondiente. Este sistema está empezando a utilizarse en Rusia y otros países.
¡Importante! Una prueba genética no puede determinar el tipo de AME. El tipo se determina en función de las capacidades funcionales del niño.
Los síntomas de la enfermedad se manifiestan en el útero en ausencia de actividad motora en el feto. Desde el nacimiento, el niño presenta hipotonía muscular generalizada con una postura característica de "rana" y la actividad motora espontánea se reduce drásticamente. No se evocan reflejos tendinosos.
Como regla general, estos niños son observados durante mucho tiempo por pediatras y neurólogos con un diagnóstico de encefalopatía perinatal. A veces los médicos asocian el complejo de síntomas de un niño letárgico con un parto difícil. Pero todos los niños con encefalopatía perinatal y las consecuencias de un parto difícil se adaptan bastante rápido y bien, mejorando gradualmente, a diferencia de los niños con AME.
El pronóstico es extremadamente desfavorable: los niños mueren, por regla general, en muy temprana edad(hasta seis meses) por enfermedades intercurrentes (que complican el curso de la enfermedad principal).
Normalmente se combinan SMA0 y SMA1.
En la atrofia muscular espinal tipo I (tipo Werdnig-Hoffmann), es posible que la madre ya note movimientos fetales tardíos y débiles durante el embarazo. Desde el nacimiento, el niño presenta una disminución generalizada del tono muscular (síndrome del "bebé fláccido"). Desde los primeros meses de vida aparece debilidad y atrofia de los músculos de las extremidades superiores e inferiores, seguida de afectación de los músculos del tronco y del cuello. Estos cambios musculares hacen que los niños no puedan sentarse. La atrofia muscular y las contracciones de las fibras musculares suelen estar enmascaradas por una grasa subcutánea bien definida. Un síntoma característico es un ligero temblor (temblor) de los dedos de las manos extendidas. A veces se detectan contracciones de los músculos de la lengua.
Un síntoma típico es también el debilitamiento o la desaparición completa de los reflejos tendinosos (rodilla, Aquiles), limitación de la movilidad normal de la articulación y deformidades esqueléticas. Debido a la debilidad de los músculos intercostales, el pecho del niño se aplana. Dado que, como resultado de la debilidad muscular, no hay suficiente ventilación de los pulmones, se producen frecuentes infecciones respiratorias y se producen diversos trastornos respiratorios. El desarrollo mental de los niños no se ve afectado.
Los bebés pueden experimentar dificultades para respirar e incapacidad para comer. Las contracturas congénitas, cuya gravedad varía desde un simple pie zambo hasta artrogriposis generalizada (múltiples patologías congénitas del aparato de movimiento, manifestadas por numerosas contracturas articulares, atrofia muscular y daño de la médula espinal), ocurren en aproximadamente el 10% de los recién nacidos gravemente afectados. Los bebés se encuentran en una posición relajada de "rana", la actividad motora espontánea se reduce, los niños no pueden superar la fuerza de gravedad de sus extremidades y no pueden mantener bien la cabeza erguida.
Por lo general, excepto en casos muy raros y graves, el diagnóstico de AME no se realiza en el hospital de maternidad. El niño es dado de alta sano y cuando los padres notan un tono muscular bajo, o no le dan mucha importancia si no saben cómo debe moverse un bebé sano, o los tranquiliza el consejo del médico de la clínica: "No te preocupes, todo el mundo se desarrolla a su debido tiempo, todo mejorará pronto" y él correrá." Es posible que los padres no comprendan la gravedad de los problemas; esto debería ser visto por un médico, pero, desafortunadamente, en las clínicas los síntomas de la AME son poco conocidos.
Estos niños muestran los primeros signos de la enfermedad antes de los 6 meses de edad. Esto significa que no adquieren las habilidades para sentarse, gatear o caminar de forma independiente. Como resultado, el rango de movimiento en estos niños es muy pequeño. Al mismo tiempo, la AME no afecta la esfera cognitiva, los niños entienden todo y la sensibilidad no se ve afectada. Si los trata como a niños normales (juegan, leen, coleccionan pirámides), entonces estos niños se desarrollan mental y psicológicamente con absoluta normalidad, y todos los retrasos en el desarrollo que los neurólogos pueden diagnosticar están asociados con una actividad motora alterada y las condiciones en las que se encuentran.
Más de 2/3 de los niños con esta enfermedad mueren antes de los 2 años; en muchos casos, la muerte ocurre en la primera infancia debido al daño a los músculos respiratorios y la aparición de diversas complicaciones en los pulmones.
En la atrofia muscular espinal tipo II, la enfermedad aparece algo más tarde (en el primer año y medio de vida del niño) y se caracteriza por un curso más lento. El síntoma principal es la incapacidad del niño para mantenerse en pie.
Los niños con AME2 generalmente pueden succionar y tragar, y la función respiratoria no se ve afectada en la primera infancia. El tono de voz nasal y la dificultad para tragar aparecen a mayor edad. A pesar de la debilidad muscular progresiva, muchos de ellos sobreviven hasta la edad escolar y más allá, aunque en las últimas etapas de la enfermedad hay un grado severo de discapacidad y los niños necesitan sillas de ruedas. Los cochecitos convencionales no son adecuados para ellos, se necesitan soportes, topes y dispositivos especiales adicionales para fijar de manera óptima la posición del cuerpo.
En muchos pacientes con una larga esperanza de vida, una de las principales complicaciones de la enfermedad son la escoliosis y las contracturas, que se desarrollan muy rápidamente. Incluso en los niños postrados en cama, la escoliosis se desarrolla de manera bastante significativa; la curvatura de la columna no se produce por carga, sino por debilidad.
Los niños con AME2 son bastante estables durante un período de tiempo y los padres pueden apoyar el conjunto de habilidades que estos niños ya han adquirido.
Existe el término "meseta de la enfermedad", que se refiere a un cierto período durante el cual los niños están bastante estables y no hay un deterioro significativo en la condición o progresión de la enfermedad. Puede verse así: los niños adquieren habilidades como todos los niños normales, cuando "empiezan" la enfermedad deja de desarrollarse y luego su condición puede retroceder muy lentamente o, por el contrario, muy rápidamente, para cada niño a su manera. O esto: los niños adquieren habilidades durante un tiempo determinado, luego ocurre algún evento o enfermedad, algunas de las habilidades se pierden y luego ocurre una "meseta" bastante larga, durante la cual puede haber incluso mejoras menores, pero luego ocurre un deterioro inevitable. La tasa de progresión de la enfermedad, la duración de la meseta (o la ausencia de ella) y el deterioro posterior son todos muy individuales.
La enfermedad de Kugelberg-Welander es la forma más leve de atrofia muscular espinal (AME tipo III). Comienza entre las edades de 1,5 y 17 años. Las personas viven más tiempo con esta enfermedad y la progresión es lenta. La atrofia muscular comienza en las piernas y luego se extiende a los brazos. En la infancia, las manifestaciones clínicas de la enfermedad pueden estar ausentes. Se desarrolla debilidad progresiva en las extremidades proximales, especialmente en los músculos de la cintura escapular. Los pacientes conservan la capacidad de caminar de forma independiente. Rara vez aparecen síntomas de debilidad de los músculos bulbares. Aproximadamente el 25% de los pacientes con esta forma de AME tienen hipertrofia muscular en lugar de atrofia muscular; por lo tanto, la distrofia muscular puede diagnosticarse erróneamente. Los pacientes pueden sobrevivir hasta la edad adulta.
La AME en este gran grupo de niños se detecta a la edad de más de un año y medio, es decir, Normalmente el niño puede caminar solo. La enfermedad se manifiesta de forma tan individual que el diagnóstico se puede realizar al año y medio o a los nueve años. Dependiendo de esto, habrá diferentes previsiones tanto de duración como de calidad de vida.
En los niños con AME3, la esperanza de vida casi no difiere del estándar: viven hasta 30 y 40 años. Sus síntomas no progresan tan rápidamente, pero estos niños también necesitan recibir clases de rehabilitación y fisioterapia.
Hay suficiente ahora un gran número de Pacientes adultos con AME, y este no es el cuarto tipo, son niños con AME2 y AME3 que han crecido. Ellos tienen grandes problemas, que no están asociados con el movimiento y la respiración. No importa cuánto vivan, 20 años o 30, ya han abandonado los medicamentos porque nadie sabe qué hacer con ellos. Cada madre de un niño con AME y cada paciente adulto con AME pueden contar aproximadamente la misma historia sobre una cita con un médico: “No vengas a verme, no sabemos qué hacer contigo”; "Aún no estás muerto, ah, guau". Efectivamente, los médicos no saben qué hacer con estos pacientes y dicen: “Bueno, lo que quieres de mí es que no me traten, pero la verdad es que no sé qué hacer contigo”. Nadie trata con ellos; después de 18 años ni siquiera saben qué hacer con ellos.
Estos pacientes (a menudo pacientes invisibles porque se sientan en casa) no creen en la atención médica porque han sido dejados de lado durante toda su vida. Y sólo buscan ayuda cuando ya no es posible ignorar los síntomas, cuando el dolor es intenso, es decir. demasiado tarde, casi ya en su lecho de muerte. Hasta hace poco no se trabajaba con esta categoría de pacientes. Ahora existe la oportunidad de mejorar de alguna manera la situación, las fundaciones caritativas brindan más ayuda y se presta más atención a este problema.
La enfermedad SMA3 no se desarrolla muy rápidamente; gradualmente se produce un debilitamiento general del cuerpo y una lenta pérdida de las habilidades adquiridas.
El cuarto tipo de AME se presenta en personas mayores de 25 años. La enfermedad se desarrolla con bastante lentitud y prácticamente no afecta la esperanza de vida. Con SMA4, se pueden desarrollar temblores y puede ocurrir un debilitamiento general, incluida la fuerza muscular. Con el tiempo, SMA4 puede provocar la pérdida de la capacidad de moverse de forma independiente.
Enfermedad progresiva del sistema nervioso: atrofia muscular espinal
Es extremadamente importante que cada persona mantenga su independencia y actividad. Sin embargo, existen enfermedades en las que los pacientes van quedando gradualmente incapacitados y sólo pueden moverse con ayuda o en sillas de ruedas. Este tipo de enfermedades incluyen la atrofia muscular espinal, en la que una persona no sólo puede dejar de caminar, sino que a veces incluso no puede respirar por sí misma.
Descripción de la enfermedad.
La atrofia muscular espinal (AME, amiotrofia espinal) es un grupo completo enfermedades hereditarias Se caracteriza por la degeneración de las neuronas motoras de la médula espinal.
Esta es una de las patologías más comunes entre los trastornos genéticos. La tasa de incidencia entre los recién nacidos es de un caso por cada 6.000 a 10.000 niños, según el país estudiado. Casi la mitad de los que nacen con AME no pueden ni siquiera cumplir los dos años y mueren.
Sin embargo, la atrofia muscular no es sólo una enfermedad infantil; puede afectar a personas de todas las edades. Los científicos han descubierto que cada 50 habitantes de la Tierra es portador del gen recesivo SMN1 (neurona motora de supervivencia), que conduce a la AME. Aunque todos los tipos de esta enfermedad surgen de una mutación en una única región cromosómica, presenta muchas formas con distintos grados de síntomas en todas las edades. A pesar de la pérdida de la actividad motora de los músculos, su sensibilidad se mantiene. La inteligencia de los pacientes no se ve afectada por el proceso, es completamente normal.
Esta enfermedad fue descrita por primera vez en 1891 por Guido Werding, quien no sólo registró los síntomas, sino que también estudió los cambios morfológicos en los músculos, los nervios y la médula espinal.
Los tipos más comunes de AME son los proximales (aproximadamente el 80-90% de todos los casos), en los que los músculos ubicados más cerca del centro del cuerpo (femoral, vertebral, intercostal, etc.) son los más afectados.
Vídeo sobre un paciente con atrofia muscular espinal
Tipos de enfermedad y gravedad de las manifestaciones.
Entre los tipos proximales se distinguen cuatro formas de la enfermedad, que se agrupan según la edad de inicio del proceso, la gravedad de los síntomas y la esperanza media de vida.
Formas de amiotrofia espinal proximal - tabla
- la forma de AME con el pronóstico menos favorable y que con mayor frecuencia termina en la muerte;
- los pacientes no pueden sentarse, pararse, experimentan grandes complicaciones al alimentarse, ya que les resulta difícil tragar y succionar;
- la respiración se ve afectada.
- los niños no pueden moverse de forma independiente;
- los pacientes se sientan y comen normalmente;
- La esperanza de vida depende en gran medida del estado de los músculos respiratorios.
- enfermedad de Kugelberg-Welander;
- AME infantil crónica.
- este tipo es el que tiene menos probabilidades de causar la muerte en los niños;
- En muchos casos, el paciente puede ponerse de pie, pero experimenta una debilidad muscular grave y, a menudo, utiliza una silla de ruedas para desplazarse.
- Amiotrofia bulboespinal de Kennedy;
- AME escápulo-peroneal vulpiana.
- en casos raros, afecta la esperanza de vida;
- El debilitamiento gradual de los músculos provoca discapacidad e incapacidad para caminar de forma independiente.
Hay una serie de atrofias musculares espinales que afectan las partes distales del cuerpo. La mayoría de las veces se ven afectadas las extremidades superiores y la enfermedad en sí suele registrarse a una edad bastante adulta.
Además de esta clasificación, existe una división de la AME en formas aisladas (que ocurren solo debido al daño de las neuronas motoras en la médula espinal) y formas combinadas (la amiotrofia espinal se acompaña de enfermedades adicionales como enfermedades cardíacas, retraso mental, fracturas congénitas, etc.).
Causas y factores de patología.
Las distrofias musculares espinales se producen debido a genes recesivos heredados ubicados en el quinto cromosoma (SMN, NAIP, H4F5, BTF2p44). Como regla general, los padres no presentan manifestaciones de AME, pero ambos son portadores y en el 25% de los casos transmiten a sus hijos un gen defectuoso que altera la síntesis de la proteína SMN, lo que conduce a la degeneración de las neuronas motoras en la columna. cable.
En una determinada etapa del desarrollo embrionario, existe un programa programable. muerte celular Los precursores de las neuronas motoras se forman en exceso. De ellas, aproximadamente la mitad debería permanecer normal, que posteriormente se diferenciará en células nerviosas. Sin embargo, en un cierto período, este proceso se detiene por el funcionamiento del gen SMN. Cuando se produce una mutación, su funcionamiento se ve interrumpido y la muerte celular continúa incluso después del nacimiento del niño, lo que conduce a la atrofia muscular espinal.
La enfermedad afecta a las neuronas motoras de los cuernos anteriores de la médula espinal.
Síntomas en niños y adultos.
El síntoma principal de la enfermedad de AME es la flacidez, debilidad y atrofia muscular. Sin embargo, cada forma de amiotrofia espinal tiene sus propias características:
- En la enfermedad de Werding-Hoffman, los primeros síntomas se pueden detectar durante el embarazo mediante una ecografía, ya que el feto se mueve muy poco. Después del nacimiento, el niño no puede mantener la cabeza erguida, darse la vuelta y luego sentarse. Casi todo el tiempo el bebé yace en una posición relajada boca arriba, sin poder juntar piernas y brazos. También son frecuentes los problemas con la alimentación, ya que el bebé tiene dificultades para tragar. La respiración a menudo se ve afectada debido a la atrofia de los músculos de las costillas. Casi el 70% de los niños mueren antes de cumplir los dos años. Después del diagnóstico, se revela una formación insuficiente de los cuernos anteriores de la médula espinal. Si el paciente vive entre 7 y 10 años, la gravedad de la atrofia muscular aumenta y muere por insuficiencia cardíaca o pulmonar aguda o por problemas digestivos. En casos raros, los pacientes viven hasta 30 años, y sólo con una aparición más tardía de los síntomas (aproximadamente 2 años).
- En el segundo tipo de atrofia muscular espinal, el niño a menudo puede respirar y tragar alimentos por sí solo. Sin embargo, con el tiempo, el proceso avanza y, a mayor edad, los niños se ven confinados a sillas de ruedas. Por lo general, los padres comienzan a notar que el niño a menudo tropieza, cae y se le doblan las rodillas. La incapacidad gradual para tragar los alimentos por sí solo aparece con la edad. Además, a medida que envejece, comienza a aparecer una curvatura severa de la columna (escoliosis). Esta forma se considera relativamente benigna y permite a los pacientes vivir hasta una edad avanzada. En algunos casos, las mujeres pueden incluso tener un hijo y dar a luz, pero existe una alta probabilidad de transmitir la enfermedad por herencia. Con el cuidado adecuado y una terapia de ejercicio regular, los pacientes pueden permanecer funcionales durante mucho tiempo.
- La amiotrofia juvenil de Kugelberg-Welander se puede registrar por primera vez entre los dos y los dieciocho años de edad. De hecho Etapa temprana Puede que no haya síntomas, el niño se desarrolla completamente. Poco a poco, la debilidad comienza a aparecer en las partes proximales del cuerpo, con mayor frecuencia en los hombros y el antebrazo. Durante muchos años, el paciente puede moverse y cuidarse por sí mismo de forma independiente. A menudo se observan espasmos musculares (fasciculaciones). El pico principal de los síntomas se registra entre las edades de dos y cinco años, cuando el niño de repente tiene dificultades para correr, levantarse de la cama y subir escaleras. El curso de la enfermedad es relativamente benigno, ya que el paciente puede conservar la capacidad de moverse de forma independiente durante mucho tiempo.
- La atrofia muscular bulboespinal de Kennedy es una enfermedad ligada al sexo que se transmite en el cromosoma X y se manifiesta exclusivamente en hombres en la edad adulta. La enfermedad progresa lentamente y comienza con debilidad en los músculos de los muslos, luego, después de 10 a 15 años, se desarrollan gradualmente trastornos bulbares (daño a los nervios craneales: glosofaríngeo, vago e hipogloso). Dado que el curso de la enfermedad es extremadamente lento, prácticamente no hay tiempo para que se alteren funciones importantes y la esperanza de vida no se reduce significativamente. Muy a menudo la enfermedad se acompaña de patologías endocrinas: atrofia testicular, disminución de la libido, diabetes.
- La AME distal de Duchenne-Arana generalmente se registra entre los 18 y 20 años. Las manos son las primeras afectadas, luego todas las extremidades superiores. Durante un largo período de tiempo, la atrofia de los músculos de las piernas se produce gradualmente. En casos extremadamente raros, la enfermedad se detiene con paresia de uno de los brazos.
- La atrofia muscular espinal escapuloperonea vulpio se hace sentir por primera vez a una edad mayor (20 a 40 años). Se manifiesta como una atrofia gradual de los músculos de la cintura escapular y los extensores del pie y la pierna. El pronóstico es relativamente favorable, ya que incluso 30 años después del inicio de la enfermedad, el paciente sigue siendo capaz de moverse de forma independiente.
La AME en mujeres embarazadas se asocia con muchas complicaciones. A menudo, una mujer no puede dar a luz por sí sola y se le prescribe una cesárea.
Diagnóstico y diagnóstico diferencial.
Un método que con un 100% de probabilidad indica la presencia de atrofia muscular espinal es el análisis de ADN mediante diagnóstico genético molecular. Su objetivo es identificar un gen defectuoso en el quinto cromosoma, en el locus 5q11-q13.
Se llevan a cabo análisis bioquímicos para detectar el contenido de creatina quinasa (CPK), alanina aminotransferasa (ALT) y lactato deshidrogenasa (LDH). Si su nivel es normal, esto nos permite excluir la distrofia muscular progresiva con síntomas similares.
Utilizando EPS (investigación electrofisiológica), se registran los impulsos de la actividad bioeléctrica del cerebro y los troncos nerviosos. En la AME, se observa un ritmo en “valla” característico de las lesiones de las neuronas en los cuernos anteriores.
MRI (Imagen por resonancia magnética) y CT ( tomografía computarizada) no siempre ayudan a identificar cambios característicos de AME en las imágenes.
El diagnóstico diferencial es necesario para distinguir la atrofia muscular espinal de la distrofia muscular, la parálisis cerebral, la esclerosis lateral amiotrófica, el síndrome de Marfan, la encefalitis transmitida por garrapatas y otras enfermedades del sistema nervioso central.
Espectrometría de masas en tándem para determinar la disminución del nivel. varios aminoácidos en el cuerpo, revela una falta de proteína SMN.
Tratamiento
Por el momento, no existe un tratamiento eficaz para la atrofia muscular espinal. Tras la detección inicial de la enfermedad, está indicada la hospitalización obligatoria con diversos estudios.
Terapia de drogas
La terapia con medicamentos tiene como objetivo mejorar la conducción de los impulsos nerviosos, normalizar la circulación sanguínea y ralentizar la destrucción de las neuronas motoras. Se utilizan los siguientes medicamentos:
- Los fármacos anticolinesterásicos tienen como objetivo reducir la actividad de la enzima que descompone la acetilcolina, que, a su vez, transmite la excitación a lo largo de las fibras nerviosas. Estos incluyen Sangviritrin, Oxazil, Prozerin; Prozerin mejora el paso de los impulsos de la neurona al músculo
- suplementos biológicos que contienen L-carnitina y coenzima Q10, que mejoran el metabolismo energético en las células;
- Vitaminas B que apoyan el tono muscular normal;
- nootrópicos que estimulan el sistema nervioso central: Nootropil, Cavinton, Semax.
- Medicamentos para estimular el metabolismo en los músculos y fibras nerviosas- orotato de potasio, Actovegin, un ácido nicotínico. Actovegin mejora el metabolismo en el tejido nervioso
Dieta
Debe entenderse que la base de la nutrición de un paciente con AME debe ser alimentos que puedan proporcionar a los músculos al máximo las sustancias necesarias.
Vale la pena enriquecer la dieta del paciente con alimentos ricos en proteínas. Sin embargo, actualmente no hay datos fiables que indiquen que la condición de los pacientes mejore con una determinada dieta. En algunos casos, la ingesta excesiva de aminoácidos en el organismo puede ser perjudicial, ya que no hay suficiente tejido muscular para procesarlos.
Las legumbres son una fuente de proteínas.
Se debe reducir el contenido calórico de los alimentos, ya que debido a una actividad física insuficiente, algunos pacientes tienden a engordar.
Métodos fisioterapéuticos, incluido el masaje.
Los pacientes necesitan tener sesiones. Masaje terapéutico dirigido a mantener funciones musculares. También serán útiles la UHF (terapia de frecuencia ultraalta), la electroforesis y las prácticas manuales. hay especiales ejercicios de respiración para estimular la función pulmonar.
El masaje es un medio para tratar la amiotrofia espinal.
Usando normalizado actividad física Puede prevenir la rigidez de las articulaciones y mantener los músculos tonificados. Los ejercicios en la piscina, donde la tensión en la columna es mínima, son muy útiles. Es importante elegir los dispositivos ortopédicos adecuados que brinden soporte al pecho y las extremidades.
Remedios caseros
No existen remedios caseros para el tratamiento de la atrofia muscular espinal. Es muy importante consultar a un médico inmediatamente después de notar los primeros síntomas. Bajo ninguna circunstancia debes automedicarte, ya que esto puede ser fatal.
Pronóstico del tratamiento y posibles complicaciones.
El pronóstico del tratamiento depende en gran medida del tipo de atrofia muscular espinal. En la primera variante, más maligna, el resultado más común es la muerte prematura del paciente y, en otros casos, a veces es posible conservar la capacidad de moverse de forma independiente hasta la vejez.
Las complicaciones de la AME incluyen: escoliosis, insuficiencia pulmonar aguda, parálisis, deformidades del pecho, pérdida de las funciones de masticar y tragar.
Prevención
No existe prevención para la atrofia muscular espinal. Lo único que puedes hacer es consultar a un genetista a la hora de planificar un embarazo.
A pesar del terrible pronóstico y la incapacidad de recuperarse completamente de esta enfermedad, no debes rendirte. La atención integral y el cumplimiento de todas las recomendaciones del médico permiten en muchos casos a los pacientes vivir una vida larga e independiente.
- Imprimir
Espondilitis anquilosante: síntomas, causas.
Espondilitis anquilosante en mujeres: diferencias en los síntomas
Espondilitis anquilosante en hombres: causas, síntomas, diagnóstico
Atrofia muscular espinal tipo 1 (AME tipo 1)
Traducción de materiales del sitio web de Muscular Dyscracy UK. Original: Atrofia muscular espinal tipo 1 http://www.musculardysopathyuk.org/app/uploads/2016/05/SMA-Type-1.pdf. Al final se encuentra el archivo pdf guardado con el artículo original.
La atrofia muscular espinal (AME tipo 1) es una enfermedad neuromuscular poco común que se hereda. La AME afecta la capacidad de gatear y caminar, mover los brazos, la cabeza y el cuello, así como respirar y tragar. La AME se clasifica en tipos según la edad a la que comienzan los síntomas y los "hitos" físicos que es probable que alcance el bebé o el niño: la capacidad de sentarse, pararse o caminar.
Hay cuatro tipos principales de AME: las formas 1, 2 y 3 aparecen en la infancia. El tipo 4 aparece en la edad adulta y también se conoce como AME de inicio en la edad adulta.
Esta clasificación no es rígida. Existe un amplio espectro de gravedad entre los diferentes tipos de AME y entre niños, jóvenes y adultos dentro de cada tipo.
También existen otras formas, incluso más raras, de AME con diversas causas genéticas, incluida AME con insuficiencia respiratoria, atrofia muscular espinobulbar y AME distal.
¿Qué causa la AME?
Normalmente, el cerebro envía impulsos eléctricos a la médula espinal a lo largo de las células nerviosas hasta los músculos. Esto le permite reducirlos conscientemente y obligarlos a moverse.
La AME afecta a una gran colección de células nerviosas llamadas neuronas motoras inferiores (motoneuronas), que emergen de la médula espinal e inervan los músculos esqueléticos. Las neuronas motoras inferiores transportan impulsos nerviosos que permiten que los músculos utilizados para gatear, caminar, mover los brazos, la cabeza y el cuello, respirar y tragar, se muevan.
Para que las neuronas motoras inferiores estén sanas, el cuerpo debe producir la importante proteína SMN (Survival Motor Neuron). La capacidad del cuerpo para hacer esto está controlada por el gen SMN1 de “supervivencia de las neuronas motoras”.
Cada persona tiene dos copias del gen SMN1, una copia de cada padre. Las personas que tienen dos copias defectuosas del gen SMN1 padecen AME. Si una persona tiene una copia defectuosa del gen, es portadora. Los portadores normalmente no tienen AME ni ningún síntoma de AME. Las personas con dos copias sanas del gen no tienen AME y no son portadoras.
La AME se transmite de padres a hijos a través de los genes SMN1. Si ambos padres son portadores, su hijo puede heredar dos genes defectuosos, uno de cada padre. Si esto sucede, el niño sufrirá AME.
Tener dos genes defectuosos significa que el niño sólo puede producir pequeñas cantidades de proteína SMN. Esto da como resultado una disminución en la cantidad de neuronas motoras inferiores en la médula espinal. El impulso de la médula espinal se transmite mal a los músculos, lo que dificulta el movimiento. Los músculos no se utilizan y esto conduce a la atrofia muscular.
por conseguir información adicional Para "La genética de la atrofia muscular espinal", consulte: http://www.smasupportuk.org.uk/the-genetics-of-sma
¿Qué es la AME tipo 1?
La AME tipo 1 es la forma más desfavorable de AME. Hay alrededor del 10% de los casos de AME en la infancia. Este tipo a veces se denomina enfermedad de Werdnig-Hoffman o AME infantil aguda.
Cada niño con AME tipo 1 es diferente. Los síntomas de la AME tipo 1 suelen aparecer durante los primeros meses de vida. En algunos casos, la AME puede afectar a los bebés antes de que nazcan y las madres pueden recordar que su bebé se volvió menos activo hacia el final del embarazo.
Podemos decir que cuanto antes aparecen los síntomas de la enfermedad, más compleja es la condición del niño. Los niños más gravemente afectados mueren antes, durante o poco después del nacimiento. Estos casos a veces se denominan AME tipo 0 (cero).
A veces, los médicos indican la gravedad de la enfermedad dentro del tipo 1 utilizando clasificación decimal, por ejemplo, 1,1, 1,2, 1,5, 1,9. Si tienes dudas sobre esta clasificación, puedes contactar con el equipo médico de tu hijo.
La AME tipo 1 es una afección que no tiene cura. Aunque es imposible predecir exactamente qué sucederá, la mayoría de los niños (aproximadamente el 95%) tienen una esperanza de vida inferior a 18 meses. Por tanto, los niños a los que se les diagnostica AME durante las primeras semanas o meses tienen una esperanza de vida significativamente más corta.
¿Cómo se diagnostica la AME tipo 1?
Un médico puede hacer un diagnóstico basándose en el historial médico del niño, el examen físico y extrayendo una muestra de sangre para una prueba de ADN. La muestra se analiza para detectar la presencia de una mutación por deleción en el gen SMN1 en el cromosoma 5. Los resultados de la prueba generalmente están disponibles dentro de 2 a 4 semanas.
Si hay alguna duda sobre el diagnóstico, es posible que se necesiten pruebas adicionales, como electromiografía (EMG) o biopsia muscular, pero generalmente esto no es necesario para confirmar el diagnóstico.
¿Existe tratamiento y prevención?
Actualmente no existe un tratamiento definitivo para la AME ni medicamentos que reviertan el daño a las neuronas motoras inferiores y detengan el debilitamiento de los músculos. Sin embargo, existe un conjunto de medidas encaminadas a reducir los síntomas y mantener la calidad de vida de los pacientes durante el mayor tiempo posible.
¿Cómo se manifiesta la AME tipo 1?
EN esta sección describe los síntomas de la AME tipo 1. Es importante recordar que cada niño con AME tipo 1 tiene síntomas diferentes y la gravedad de la enfermedad varía.
Debido al tono muscular débil (hipotonía), a los niños con AME tipo 1 a menudo se les describe como "flexibles". La debilidad muscular profunda afecta la capacidad de moverse, tragar y respirar. Los bebés con esta forma de AME tienen dificultad para controlar la cabeza, darse vuelta y sentarse solos. También puede producirse un llanto leve.
La debilidad muscular en los niños suele ser igual en ambos lados del cuerpo (simétrica). Los músculos ubicados más cerca del centro del cuerpo (músculos proximales) generalmente se ven afectados con más frecuencia que los músculos ubicados más lejos del centro del cuerpo (músculos distales). Como regla general, los niños que padecen AME tipo 1 tienen más piernas débiles que las manos. Los niños tienen dificultad para levantar brazos y piernas y al mismo tiempo pueden usar las manos y los dedos.
La debilidad de los músculos respiratorios puede provocar dificultad para respirar y tos. También puede haber una mayor probabilidad de sufrir infecciones respiratorias, que pueden poner en peligro la vida.
Los músculos utilizados para chupar y tragar también se ven afectados, lo que puede provocar dificultades para alimentar y aumentar de peso al bebé. La dificultad para tragar puede aumentar el riesgo de que líquidos o alimentos entren en los pulmones (aspiración), lo que puede provocar asfixia y, en algunos casos, neumonía.
El cerebro generalmente no se ve afectado y los niños con esta forma de la enfermedad a menudo se describen como brillantes, activos y receptivos. Los músculos faciales no suelen verse afectados y los niños pueden sonreír y fruncir el ceño.
¿Qué atención médica y apoyo necesitan las personas con AME tipo 1?
el niño necesita asistencia calificada varios especialistas afines, que pueden parecer redundantes, pero cada uno tiene un papel importante que desempeñar. Estos pueden incluir especialistas en enfermedades neuromusculares, cuidados paliativos, neumólogos, ortopedistas, fisioterapeutas, terapeutas de rehabilitación, logopedas, nutricionistas y pediatras comunitarios. Es importante, si es posible, contar con un administrador de casos que pueda ayudar a coordinar los servicios brindados a la familia del paciente. Puede encontrar más información sobre el trabajo de cada especialista en la hoja informativa Quién es quién de los profesionales en: http://www.smasupportuk.org.uk/whos-who-of-professionals
En cada cita se pueden discutir cuestiones que surjan y luego decidir conjuntamente acciones futuras.
Aliento
Es necesario un control cuidadoso de la respiración para garantizar la comodidad del paciente y reducir las complicaciones de la debilidad muscular. Puede encontrar una descripción general del control de la respiración en el folleto Estándares de atención para la atrofia muscular espinal: la guía familiar publicada por TREAT-NMD. El folleto se puede obtener comunicándose con una organización de soporte de AME en el Reino Unido o descargándolo del sitio web oficial de TREAT-NMD: http://www.treat-nmd.eu/sma/care/family-guide/.
Hay varias opciones para el soporte respiratorio. Sin embargo, no todos los métodos son adecuados para todos los pacientes con AME tipo 1.
Posibles opciones:
- Fisioterapia torácica para mantener la comodidad;
- Limpiar el tracto respiratorio de secreciones;
- Tratamiento farmacológico que reduce la producción de secreciones;
- Medicamentos para el dolor para reducir la angustia causada por la dificultad para respirar;
- Ventilación no invasiva (VNI) con ventilación mecánica para aumentar la comodidad y el control del paciente. infección aguda o para corregir la hipoventilación nocturna. La ventilación no invasiva puede no ser adecuada para todos los pacientes; por ejemplo, no es adecuada para niños con debilidad muscular grave. cavidad oral y faringe (músculos bulbares), y niños menores de 6 meses. Para otros pacientes, esta opción ayudará a aliviar los síntomas de insuficiencia respiratoria o facilitará el alta del hospital a su domicilio;
- La ventilación invasiva es ventilación artificial a través de un tubo endotraqueal (un tubo de plástico flexible que se inserta a través de la boca o la nariz hasta la tráquea) o traqueotomía. La ventilación mecánica con tubo endotraqueal se utiliza a menudo como medida a corto plazo en casos de Asistencia de emergencia. Sin embargo, siempre que exista un tratamiento eficaz para prevenir la progresión de la debilidad muscular, el uso de un tubo endotraqueal para ventilación artificial sigue siendo un dilema ético a largo plazo.
Seleccionar la opción de control de la respiración más adecuada implica decisiones muy difíciles. Se necesita tiempo y apoyo para aclarar todas las cuestiones y discutir las distintas opciones que sean mejores para el niño. Las decisiones se toman conjuntamente con especialistas que conocen el historial médico del niño y pueden predecir su posible evolución.
Nutrición
El niño puede tener dificultades para alimentarse y tragar debido a la debilidad muscular. La alimentación puede ser abrumadora para los bebés con AME tipo 1 y, como resultado, pueden perder peso. Los niños con dificultad para tragar corren el riesgo de inhalar (aspirar) alimentos, lo que puede causar infecciones respiratorias (respiratorias).
Se puede obtener asesoramiento y apoyo sobre la alimentación, la deglución y la nutrición de profesionales de la salud, como cuidadores, médicos consultores, logopedas, dietistas y enfermeras comunitarias. Un terapeuta ocupacional y un fisioterapeuta también pueden recomendar cómo sostener adecuadamente a su bebé mientras lo alimenta.
Actualmente no hay evidencia de que las personas con AME tipo 1 requieran una dieta terapéutica especial o una dieta que aumente o disminuya ciertos nutrientes.
Si no es seguro tragar o el bebé no aumenta de peso, se pueden ofrecer métodos de alimentación alternativos, como la sonda nasogástrica (NGT), la sonda nasoyeyunal o la alimentación por sonda gastrointestinal.
Todos deberían tener la oportunidad de discutir las indicaciones para el uso de los métodos anteriores y considerar todos los posibles beneficios y riesgos para el niño. Independientemente de la opción elegida, es necesario brindar preparación y apoyo para que el niño pueda seguir alimentándose por sí solo y de forma segura en el hogar.
El estreñimiento es un problema común en los niños con AME tipo 1. Puede causar molestias y causar problemas respiratorios. Algunos niños experimentan reflujo. Para reducir el malestar y prevenir complicaciones, se debe discutir el manejo de estos síntomas con los proveedores de atención del niño.
Cuidado y apoyo
Debería tener la oportunidad de discutir en detalle las opciones de cuidado que son apropiadas para su hijo. Un equipo de especialistas te ayudará a decidir qué apoyo es el más adecuado en cada caso. Es importante desarrollar un plan con anticipación para tratar a su hijo en caso de deterioro o emergencia. El plan puede revisarse en cualquier momento si lo desea.
En una situación ideal, el objetivo de la atención es mejorar la calidad de vida del paciente en su domicilio con su familia durante el mayor tiempo posible, con un número mínimo de hospitalizaciones.
Además de la atención respiratoria y nutricional, hay apoyo adicional disponible para mejorar la salud del niño y el bienestar emocional de toda la familia. Mientras el niño está en casa, este apoyo lo proporciona un médico de cabecera, una enfermera o un equipo de atención sanitaria. Cuidados paliativos. [ Nota ed.: les recordamos que el artículo fue desarrollado por una organización británica] En el Reino Unido, los hospicios para niños también ofrecen una amplia gama de servicios para apoyar a los niños con enfermedades terminales y sus familias. Obtenga más información sobre los hospicios y cuidados paliativos para niños locales en http://www.togetherforshortlives.org.uk/ o llame al 100.
Para mantener la movilidad del niño se recurre a la fisioterapia para realizar ejercicios pasivos que el niño no puede realizar por sí solo. Puedes utilizar estas técnicas en casa. El ejercicio pasivo también es bueno para la circulación de su bebé y ayuda a prevenir la rigidez (contractura) de las articulaciones.
Un fisioterapeuta puede recomendarle realizar estiramientos y ejercicios a su hijo mientras se baña, nada o está en un baño de hidroterapia. Realizar estos ejercicios de forma lúdica permitirá que el niño pase tiempo con placer y moverse en agua tibia agregará una sensación de libertad de movimiento.
La fisioterapia torácica puede ayudar a despejar las vías respiratorias si tiene problemas para toser.
Elegir una posición general puede mejorar la comodidad general del bebé. El especialista en rehabilitación puede sugerirle sentar al niño, lo que le brindará el apoyo necesario y le permitirá jugar tranquilamente.
El especialista también puede recomendar el uso de sistemas para dormir (colchones) que asegurarán una posición cómoda para los brazos y piernas del niño durante la noche.
¿Qué otra ayuda es posible?
Ser diagnosticado con AME tipo 1 tiene un profundo impacto en una familia. En tal situación es muy importante contar con apoyo emocional y la oportunidad de discutir temas emergentes. Dicho apoyo puede incluir especialistas calificados, un terapeuta, un trabajador médico independiente, Trabajador social, psicólogo o psicoterapeuta.
[ Nota Ed.: Le recordamos que el artículo fue desarrollado por una organización británica] Además de la atención básica brindada por varios especialistas, la información y asistencia necesaria se puede obtener contactando al Centro de Soporte SMA en el Reino Unido (Spinal Muscular Acracy Support UK). ). Los empleados del centro responderán cualquier pregunta que pueda tener y también podrán proporcionar información de contacto para los voluntarios que tengan experiencia personal combatir esta enfermedad. En el Reino Unido, los niños con AME tipo 1 reciben juegos educativos gratuitos.
Para obtener más información, visite: http://www.smasupportuk.org.uk/how-we-can-support-you, llame al 520 o envíe un correo electrónico: .
Muscular Dystruphy UK también proporciona información, apoyo, promoción y subvenciones para proporcionar equipos a personas que padecen atrofia muscular espinal y una variedad de otras enfermedades neuromusculares. La dirección de su sitio web: www.musculardysopathyuk.org. También puedes llamar al 6352 o enviar un correo electrónico: .
EN diferentes regiones En el Reino Unido, las clínicas neuromusculares del gobierno cuentan con consultores y especialistas locales. De ellos puede obtener toda la información necesaria y ayuda sobre las enfermedades musculares. Los contactos de especialistas locales se proporcionan en el enlace: http://www.musculardystropicuk.org/get-the-right-care-and-support/people-and-places-to-helpyou/care-advisors/
Soporte financiero
Las familias que viven en el Reino Unido, dependiendo de su situación específica, pueden tener derecho a diversas prestaciones para cubrir los costes adicionales que surjan.
Puede encontrar más información sobre los beneficios en: http://www.gov.uk/ en las secciones "Beneficios" y "Cuidadores y beneficios por discapacidad". Puede comunicarse con el Departamento de Trabajo y Pensiones del Reino Unido llamando al 8545.
Contact a Family proporciona información esencial y apoyo a familias con niños discapacitados, incluida información sobre beneficios y subsidios. Puede contactarlos por teléfono: 3555 o a través del sitio web oficial: http://www.cafamily.org.uk/
Disability Rights UK publica un boletín gratuito y una Guía de derechos de las personas con discapacidad cada año. Puede encontrar más información en: http://www.disabilityrightsuk.org/
Together for Short Lives brinda información y apoyo a familias con niños que viven con enfermedades terminales. Puede ponerse en contacto con esta organización por teléfono: 100 o a través de la página web oficial: http://www.togetherforshortlives.org.uk/
Turn2Us es una organización benéfica que ayuda a las personas a recibir beneficios de seguridad social, subvenciones y otras ayudas. Puede contactar con ellos por teléfono: 2000 o a través del sitio web: http://www.turn2us.org.uk/
Para solicitar beneficios económicos, puede comunicarse con el profesional de la salud independiente con el que trabaja, enfermera del distrito, un especialista en enfermedades neuromusculares o un trabajador social.
También hay una gran cantidad de organizaciones benéficas que ayudan a comprar artículos para el hogar, equipos especializados u organizar un día libre. Para obtener más información, póngase en contacto con SMA Support UK o utilice el mapa del sitio: http://www.routemapforsma.org.uk/
Asesoramiento genetico
Los padres de niños con AME pueden recibir una derivación para una consulta sobre cuestiones hereditarias, incluso de un médico local.
Este asesoramiento lo realiza un especialista en genética. Él responderá todas las preguntas y lo ayudará a descubrir cómo se transmite la AME y cuáles son las posibilidades de desarrollar esta enfermedad en otros miembros de la familia. Un especialista en genética también puede asesorar a los futuros padres a la hora de planificar un embarazo. Puede solicitar una segunda consulta en cualquier momento.
Puede encontrar más información sobre la genética de la AME, los riesgos de transmitir la enfermedad a un niño y las pruebas necesarias en el folleto "La genética de la atrofia muscular espinal" en: http://www.smasupportuk.org.uk/ la-genética-de-sma
Puede encontrar información sobre opciones futuras durante el embarazo en: http://www.smasupportuk.org.uk/future-options-in-pregnancy
¿Qué se espera en el futuro?
Para obtener información sobre la investigación de la AME, puede visitar http://www.smasupportuk.org.uk/research
A medida que se desarrollan nuevos medicamentos, es necesario probarlos en entornos clínicos y, a veces, lleva años encontrar el número adecuado de pacientes para los ensayos.
En el Reino Unido existe el Registro de Pacientes con AME, una base de datos de información genética y clínica sobre personas con AME que desean acelerar el proceso de investigación. El registro permite a los especialistas obtener información sobre la condición y número de pacientes con esta enfermedad. Esta información contribuye al desarrollo y mejora de los estándares de atención al paciente.
Obtenga más información en: http://www.treat-nmd.org.uk/registry escribiendo a dirección de correo electrónico: http:// o llamando al 8605.
Recursos adicionales
Se pueden solicitar copias impresas de los siguientes folletos al servicio SMA Support UK llamando al 520 o descargándolos del sitio web oficial en: http://www.smasupportuk.org.uk/about-sma
- Atrofia muscular espinal - Información para las familias
- Cuidando a tu hijo
- Juguetes, juegos y actividades para niños con atrofia muscular espinal
- ¿Quién es cuál de los especialistas?
- Genética de la atrofia muscular espinal
- Posibles opciones durante el embarazo.
- Información y soporte
- Servicio de Asistencia Social
Estándares para la atención y tratamiento de la AME (TREAT-NMD)
[ Nota TREAT-NMD es una organización europea dedicada a las enfermedades neuromusculares. ]
Este folleto describe las mejores prácticas para controlar y tratar las formas más comunes de AME, incluida la AME tipo 1. Lo utilizan los médicos pero también está disponible para las familias. Se puede solicitar una copia impresa a la organización benéfica SMA Support UK. También se puede descargar desde la página web oficial de la organización TREAT-NMD: www.treat-nmd.eu/sma/care/family-guide/.
Registro de pacientes con AME del Reino Unido
El Registro de Pacientes es una base de datos de información genética y clínica sobre personas con AME. Se utiliza para encontrar participantes para ensayos clínicos y también para ayudar a los especialistas a obtener más información sobre una enfermedad. Puede obtener información sobre el funcionamiento del Registro y cómo registrarse en él en la organización benéfica británica SMA Support UK en: www.treat-nmd.org.uk/registry. También se puede contactar con la organización a través del teléfono: 8605.
Glosario
Aminoácidos
Unidad básica de la que están hechas las proteínas. Hay 20 aminoácidos diferentes que intervienen en la formación de compuestos proteicos. El orden específico de los aminoácidos determina la estructura y función de una proteína.
Amniocentesis
Recolectar una muestra de líquido amniótico (el líquido que contiene al feto) para diagnóstico prenatal. Las células del líquido se analizan para detectar posibles trastornos genéticos.
Líquido amniótico
El líquido que rodea al feto en el útero.
Bocina delantera
La porción anterior de la médula espinal, que contiene los cuerpos celulares de las neuronas motoras inferiores. Proyecciones largas y delgadas de neuronas motoras llamadas axones transportan impulsos desde el asta anterior de la médula espinal hasta los músculos.
Anticuerpos
Proteínas producidas por el organismo para protegerlo de cuerpos extraños como bacterias o virus.
Aspiración
Ingestión de alimentos, líquidos o vómitos en las vías respiratorias/pulmones.
Atrofia
Agotamiento o reducción de cualquier órgano. La AME se llama atrofia muscular espinal debido al daño a las neuronas motoras inferiores dentro de la médula espinal, lo que conduce a la atrofia de los músculos esqueléticos.
Herencia autosómica recesiva
Para que se herede una enfermedad genética, ambos padres deben ser portadores de un gen recesivo (suprimido), una copia dañada del gen de cada uno. Si una persona tiene sólo una copia defectuosa de un gen, normalmente no muestra síntomas de la enfermedad, pero es portador y puede transmitir el gen dañado a sus hijos. La enfermedad es autosómica si el gen defectuoso se localiza en el autosoma. La AME suele ser un trastorno autosómico recesivo.
autosoma
Cualquiera de los 22 pares de cromosomas en cuerpo humano, que no participan en la determinación del sexo. Son idénticos en hombres y mujeres. Cada par de autosomas (uno del padre y otro de la madre) contiene genes para los mismos rasgos característicos.
axón
Un proceso alargado y delgado de una célula nerviosa. Los axones transportan impulsos eléctricos desde el cuerpo celular (donde se encuentra el núcleo) hasta su objetivo, como los músculos.
Músculos bulbares
Músculos alrededor de la boca y la garganta. Cuando estos músculos se ven afectados, puede resultar difícil tragar y hablar.
Dióxido de carbono
Gas que se produce como uno de los productos finales cuando una célula utiliza oxígeno para producir energía. Se libera con el aire exhalado a través de los pulmones.
Transportador
Este término se refiere a patrones de herencia autosómicos recesivos y recesivos ligados al cromosoma X. Una persona que tiene una copia defectuosa y una copia sana del gen es portadora. Normalmente, debido a la presencia de una copia sana del gen, los portadores no presentan ningún síntoma, pero pueden transmitir la enfermedad a sus hijos. En la AME, los portadores tienen una copia defectuosa del gen de supervivencia de la neurona motora 1 (SMN1) y una copia sana de SMN1. Dos portadores de la mutación del gen SMN1 tienen un 25% (1 en 4) de posibilidades de tener un hijo con AME por cada embarazo. Un niño debe heredar dos copias del gen SMN1 defectuoso de cada padre para poder desarrollar AME.
Celúla
La unidad estructural más simple de un organismo vivo. Las células son de diferentes tipos, como neuronas motoras (un tipo de célula nerviosa), queratinocitos (células epidérmicas) o eritrocitos (glóbulos rojos).
Sistema nervioso central (SNC)
El sistema nervioso central está formado por el cerebro y la médula espinal. El SNC se conecta con otros órganos y tejidos del cuerpo, como los músculos esqueléticos, a través del sistema nervioso periférico (SNP).
Obtención de muestras de vellosidades coriónicas.
Obtener una muestra de vellosidades coriónicas es una forma de comprobar si un feto tiene AME. Se obtiene una muestra de células vellosas coriónicas (tejido placentario) utilizando una aguja. Este procedimiento suele realizarse entre la undécima y decimocuarta semana de embarazo. De esta manera, se pueden analizar genéticamente las células para detectar la presencia de AME.
cromosomas
Un cromosoma es una estructura que contiene ADN. Cada célula humana contiene 46 cromosomas (con algunas excepciones, incluidos los espermatozoides y los óvulos). Heredan 23 de su madre y 23 de su padre, formando 23 parejas.
Ensayo clínico
Ensayo realizado en personas para probar un tratamiento o una intervención para obtener más información sobre una enfermedad.
contractura
Estrechez en el tejido conectivo y los tendones alrededor de una articulación, lo que provoca debilidad e incapacidad para flexionar y extender completamente la articulación.
Supresión
Material genético (parte del ADN) que falta en un cromosoma o gen.
Diagnóstico
Detección de la enfermedad por síntomas o mediante pruebas genéticas. Se realiza un diagnóstico clínico cuando el médico ve suficientes síntomas para estar seguro de que el paciente tiene la enfermedad sospechada. Cuando se trata de enfermedades genéticas, el diagnóstico se confirma luego de realizar una prueba genética y luego de encontrar el gen dañado que causa la enfermedad. Los médicos expertos en el campo de la AME suelen diagnosticar este tipo de enfermedades con un alto grado de precisión, basándose en síntomas clínicos. Sin embargo, generalmente se recomienda realizar pruebas genéticas para todos los trastornos genéticos para garantizar que se proporcione el tratamiento correcto y garantizar que la familia pueda beneficiarse de las pruebas prenatales en el futuro si lo desea.
distal
Término anatómico que significa la ubicación más alejada del centro del cuerpo hacia las extremidades. Los músculos distales, como los de los brazos y las piernas, tienden a verse menos afectados por las formas más comunes de AME en comparación con los músculos proximales, los que participan en la respiración.
ADN (ácido desoxirribonucleico)
El ADN es una molécula que contiene el modelo genético para el desarrollo de todos los organismos conocidos. El ADN a menudo se compara con un conjunto de planos, una receta o un código porque contiene las instrucciones necesarias para producir otros componentes de las células, como las proteínas.
Electromiograma (EMG)
Una prueba que evalúa la actividad eléctrica de los músculos y los nervios que controlan los músculos. Se utiliza para diagnosticar enfermedades neuromusculares. Hay dos tipos de EMG: intramuscular y superficial. La EMG intramuscular implica insertar un electrodo de aguja, o una aguja que contiene dos electrodos de alambre fino, a través de la piel hasta el músculo. La EMG de superficie implica colocar un electrodo en la superficie de la piel.
Embrión
Nombre que corresponde a la etapa de desarrollo desde el óvulo fecundado hasta las ocho semanas de gestación, cuando el embrión se convierte en feto.
Enzima
Proteína que inicia, promueve o acelera una reacción química. Casi todos los procesos que ocurren en nuestro cuerpo requieren enzimas. Por ejemplo, digestión de alimentos, crecimiento y estructura celular.
Término utilizado para un feto después de la octava semana de desarrollo hasta que nace.
Tubo gastronómico (tubo G)
Una sonda de alimentación colocada quirúrgicamente en el estómago. A veces, el procedimiento de colocación de la sonda también se denomina PEG (gastrostomía endoscópica percutánea).
Sección de ADN que transporta información sobre la síntesis de proteínas. Los genes son portadores de la herencia de una generación a la siguiente. Normalmente tenemos dos copias de cada gen, heredadas de cada padre. Cuando los genes mutan, la estructura y función de las proteínas cambian, lo que puede provocar enfermedades.
Asesoramiento genetico
Información y apoyo brindado por un especialista en genética a personas con antecedentes familiares de trastornos genéticos. El asesoramiento genético ayuda a las familias a comprender cómo se transmite la enfermedad, la probabilidad de transmitirla a sus hijos y qué miembros de la familia pueden ser portadores del gen dañado. El asesoramiento también ayuda a los adolescentes/jóvenes con esta afección a comprender cuáles son sus opciones para el futuro.
Desordenes genéticos
Enfermedades causadas por cambios genéticos. Los trastornos genéticos pueden ser causados por daño a uno o más genes, o incluso a cromosomas completos.
Prueba genética
Estudio de los genes de una persona para identificar cambios que puedan causar enfermedades genéticas.
Genética
Herencia
La transmisión de rasgos (características) mediante la herencia de genes de una generación a otra.
hipotensión
Tono muscular disminuido/bajo, a veces descrito como flacidez.
hipoventilación
Disminución de la frecuencia y profundidad de la respiración (demasiado superficial o demasiado lenta), lo que provoca un aumento de dióxido de carbono en el cuerpo.
Intubación
Procedimiento para insertar un tubo a través de la boca o la nariz hasta la vía respiratoria principal (tráquea) para Respiración artificial. La intubación se puede realizar como parte de un procedimiento electivo, p. cirugía para protección respiratoria, o en caso de emergencia si el paciente se encuentra en estado crítico.
Ventilación invasiva
Este término describe la asistencia respiratoria que se suministra al cuerpo a través de un dispositivo o tubo. Esto generalmente requiere intubación o traqueotomía. Esto se diferencia de la ventilación no invasiva, que se realiza mediante una mascarilla o una boquilla de respirador.
Ventilación artificial
Un procedimiento médico que se utiliza para ayudar con la respiración cuando un paciente no puede respirar por sí solo. Generalmente se realiza utilizando un ventilador especial o una bolsa de compresión manual. El término se utiliza con mayor frecuencia para referirse a formas invasivas de ventilación, como la intubación o la traqueotomía. Sin embargo, en algunos casos, la ventilación mecánica a corto plazo puede ser no invasiva.
Molécula
Dos o más átomos unidos químicamente entre sí. Por ejemplo, el agua es una molécula formada por dos átomos de hidrógeno y un átomo de oxígeno unidos entre sí (H2O).
Neuronas motoras (neuronas motoras)
Células nerviosas que conectan el cerebro y la médula espinal con los músculos esqueléticos, permitiendo la contracción (movimiento) muscular consciente. Actúan como un sistema de transmisión de mensajes: los impulsos eléctricos que se originan en el cerebro se transmiten a lo largo de la médula espinal a través de las neuronas motoras superiores; Luego, los impulsos eléctricos se transmiten a través de las neuronas motoras inferiores a los músculos esqueléticos, que controlan el movimiento. Las neuronas motoras inferiores están ubicadas en el asta anterior de la médula espinal y son las principales células afectadas por la AME. En la AME, cantidades insuficientes de proteína SMN provocan daño a las neuronas motoras inferiores, lo que conduce aún más a debilidad y atrofia muscular.
Biopsia muscular
Tomar una muestra de tejido muscular para investigación.
Mutación
Un cambio irreversible en la secuencia de ADN de un gen que puede ser heredado por generaciones posteriores. Dependiendo del tipo de mutación y su ubicación dentro de un gen, puede no tener ningún efecto sobre la producción de proteínas o dañar la función de producción de proteínas, causando enfermedades genéticas como la AME.
Sonda nasogástrica (NG)
Una sonda de alimentación delgada y flexible que pasa a través de la nariz. El extremo del tubo está en el estómago.
tubo nasoyeyunal
Un tubo de alimentación delgado y flexible que se inserta en la nariz. El extremo del tubo está en yeyuno(intestino delgado medio).
Células nerviosas
A menudo llamadas neuronas, las células nerviosas transmiten rápidamente impulsos eléctricos por todo el cuerpo. Diferentes tipos de células nerviosas forman el sistema nervioso, que nos permite percibir y responder a ambiente. Por ejemplo, el cerebro envía un impulso a las células nerviosas, provocando que se contraigan. Las células nerviosas desempeñan un papel importante tanto en funciones inconscientes, como los latidos del corazón, como en funciones conscientes, como el movimiento de las manos.
neuromuscular
Todo lo que tenga que ver con nervios, músculos o uniones neuromusculares.
Unión neuromuscular (UNM)
Una conexión entre las neuronas motoras inferiores y las fibras del músculo esquelético llamada sinapsis. La NMJ permite que los impulsos nerviosos se transmitan a los músculos, provocando que se contraigan.
Ventilación no invasiva (NVL)
Soporte respiratorio mediante una máquina que suministra aire a través de una mascarilla.
El centro principal de la célula, que contiene moléculas de ADN.
Terapia de rehabilitación
Observación y tratamiento para desarrollar habilidades para la vida independiente.
Ortopédico
Perteneciente al sistema musculoesquelético: músculos y esqueleto, incluidas articulaciones, ligamentos, tendones y nervios.
Cuidados paliativos
Los cuidados paliativos son cuidados completos del cuerpo, la mente y el alma del paciente, así como el apoyo a la familia del paciente. La ayuda se brinda ya en la etapa de diagnóstico de la enfermedad y continúa independientemente de si el paciente recibe tratamiento o no (Definición de la Organización Mundial de la Salud, 1998). Los cuidados paliativos se pueden brindar en una variedad de entornos, incluidos hospitales y hospicios, y en el hogar.
PEG (gastrostomía endoscópica percutánea)
Una sonda de alimentación que se coloca en el estómago a través de la pared abdominal. El tubo se coloca mediante una cámara endoscópica especial. En algunos casos, el procedimiento se realiza utilizando sedantes sin anestesia general.
Sistema nervioso periférico (SNP)
Consiste en procesos de células nerviosas ubicadas fuera del sistema nervioso central (SNC). El SNP conecta el sistema nervioso central con músculos y órganos internos. Los axones de las neuronas motoras inferiores y sus conexiones con los músculos (uniones neuromusculares) se encuentran dentro del SNP.
Fisioterapia
Métodos físicos utilizados para fortalecer, mantener y restaurar. función física cuerpo.
Diagnóstico prenatal
Pruebas genéticas para detectar la presencia de enfermedades en el feto o embrión. Se realizan procedimientos como la amniocentesis o la biopsia de vellosidades coriónicas para extraer muestras de líquido o tejido.
Proteína
Las proteínas están formadas por cadenas de aminoácidos dispuestos en en un cierto orden. El orden de los aminoácidos en la cadena está determinado por el código genético (ADN). Varios genes tienen "instrucciones" para la síntesis de proteínas. Las proteínas son material de construcción nuestro cuerpo y son necesarios para la estructura, funcionamiento y regulación de células, tejidos y órganos. Ejemplos de diferentes proteínas son las enzimas, las hormonas, los anticuerpos y la proteína de la neurona motora de supervivencia (SMN).
proximal
Término anatómico que significa una ubicación más cercana al centro del cuerpo. Los músculos proximales, como los que se encuentran en las caderas, los hombros y el cuello, se ven más afectados que los músculos distales en la mayoría de los casos de AME.
Enfermedad rara
La Unión Europea (UE) considera que una enfermedad es rara si afecta a menos de cinco personas.
Recesivo
Autosómico recesivo es el patrón de herencia de una enfermedad genética en presencia de dos copias defectuosas del gen. Esto significa que se hereda una copia defectuosa del gen de cada padre. La AME, causada por una mutación en el gen de supervivencia de la neurona motora 1 (SMN1), es un trastorno autosómico recesivo. En las enfermedades recesivas ligadas al cromosoma X, se requieren dos copias defectuosas para que la enfermedad genética se manifieste en las mujeres, pero sólo se requiere una copia del gen defectuoso para que la enfermedad se manifieste en los hombres. Esto ocurre porque las enfermedades recesivas ligadas al cromosoma X son causadas por mutaciones en genes del cromosoma X pero ausentes en el cromosoma Y. Los hombres tienen un cromosoma X y un cromosoma Y, mientras que las mujeres tienen dos cromosomas X.
Reflujo
El reflujo de líquido desde el estómago hacia el esófago.
Respiratorio
Relativo a la respiración.
ARN (ácido ribonucleico)
El ARN es muy similar al ADN en que también transporta información genética. Desempeña un papel importante en la formación de proteínas. Hay diferentes tipos de ARN que desempeñan diferentes funciones.
Músculo esquelético
Un músculo controlado conscientemente unido a los huesos que permite el movimiento. Por ejemplo, los músculos bíceps, tríceps y muslos.
Espinal
Relativo a la columna vertebral.
Médula espinal
Un haz de tejido nervioso dentro de la columna. Incluye células nerviosas y se extiende desde el cerebro. El cerebro y la médula espinal forman el sistema nervioso central (SNC).
Gen de supervivencia de la neurona motora 1 (SMN1)
Un gen que, cuando se muta o elimina, causa AME. Para que las neuronas motoras inferiores sobrevivan, se necesita una cierta cantidad de proteína SMN, que es producida por el gen SMN1.
Gen de supervivencia de la neurona motora 2 (SMN2)
Gen que influye en la gravedad de la AME porque es capaz de producir pequeñas cantidades de la proteína SMN. En personas con un gen SMN1 defectuoso, es importante tener más copias del gen SMN2, porque cuantas más copias del gen SMN2 tenga una persona, más podrá el cuerpo producir proteína SMN funcional. Los pacientes con formas más graves de AME, como los tipos 1 y 2, suelen tener menos copias del gen SMN2 que los pacientes con AME tipo 3.
Gen de supervivencia de la neurona motora (SMN)
El gen que produce la proteína (SMN). Las mutaciones en el gen SMN1 son la causa de algunas formas de AME. Hay dos tipos de genes SMN: SMN1 y SMN2.
proteína SMN
Producida a partir de los genes SMN1 y SMN2, la proteína SMN es necesaria para la supervivencia de las neuronas motoras inferiores. Si una célula carece de la proteína SMN, muere. De todos los tipos de células, las neuronas motoras inferiores son las más afectadas por niveles bajos de proteína SMN.
Simétrico
Lo mismo en ambos lados.
Textil
Un sistema de células que funcionan simultáneamente. Por ejemplo, los órganos se forman a partir de varios tejidos.
Tráquea
Traqueotomía
Cirugía para crear una abertura en la tráquea que permita respirar a través de un tubo en lugar de por la boca.
Respirador
Dispositivo de ventilación mecánica.
Virus
Los virus están compuestos de material genético (ADN o ARN) rodeado por una cubierta proteica. Son capaces de adherirse a las células y penetrar en su interior. Algunos virus (como los del resfriado y la gripe) enferman a las personas. Pero la capacidad de penetrar en las células también significa que algunos virus pueden usarse para tratamiento.
Traducción de Ekaterina Firsova.